Документ Microsoft Office Word (3). Обзор статей журнала Фармация по фармацевтическому анализу за 2002 год
 Скачать 1.23 Mb. Скачать 1.23 Mb.
|

|
ГОСУДАРСТВЕННОЕ БЮДЖЕТНОЕ ОБРАЗОВАТЕЛЬНОЕ УЧРЕЖДЕНИЕ ВЫСШЕГО ПРОФЕССИОНАЛЬНОГО ОБРАЗОВАНИЯ «БАШКИРСКИЙ ГОСУДАРСТВЕННЫЙ МЕДИЦИНСКИЙ УНИВЕРСИТЕ МИНИСТЕРСТВА ЗДРАВООХРАНЕНИЯ РОССИЙСКОЙ ФЕДЕРАЦИИ Кафедра фармацевтической химии с курсами аналитической и токсикологической химии Тема: «Обзор статей журнала «Фармация» по фармацевтическому анализу за 2002 год» Курсовая работа студентки фармацевтического факультета заочного отделения группы 503 А Шайбаковой (Минликаевой) Юлии Леонидовны Руководитель: зав. кафедрой, профессор Халиуллин Ф.А. Уфа - 2015 Содержание
Введение Цель курсовой работы - изучение применения методов в фармацевтическом анализе лекарственных препаратов. Задачи курсовой работы: 1.Провести литературный обзор статей из журнала «Фармация» за 2002 год по фармацевтическому анализу; 2. Описать методики определения различных веществ
Арника горная — Arnica montana L., сем. Asteraceae (сложно-цветные) — относится к так называемым полихрестам, т.е. является растением, издавна применяемым как в аллопатии (официальной медицине), так и гомеопатии. Действие гомеопатических препаратов арники обусловлено комплексом биологически активных веществ: сесквитерпеновыми лактонами, фенолкарбоновыми кислотами, дубильными веществами, углеводами, аминокислотами и т.д. Известно, что некоторые свободные аминокислоты оказывают положительное влияние на сердечно-сосудистую систему, участвуют в процессах нервной регуляции различных функций организма, а также влияют на сосудистый тонус. В гомеопатии в качестве лекарственного растительного сырья используют высушенные цветки, цветущую надземную часть, корневища с корнями и целое цветущее растение арники горной в свежем виде. При изучении компонентного состава биологически активных веществ сухих цветков арники горной методом хроматографии на бумаге выявлены цистеин, лейцин, аспарагиновая кислота, гистидин, метионин, триптофан, причем гистидин и аспарагиновая кислота — в значительных количествах, а цистеин, лейцин, метионин и триптофан — в следовых. В подземной части арники горной обнаружена пипеколиновая кислота Цель настоящего исследования — изучение компонентного состава аминокислот в гомеопатических матричных настойках арники горной, полученных из целого цветущего свежего растения, а также из высушенных корневищ с корнями. Экспериментальная часть Объектами исследования служили гомеопатические матричные настойки арники горной, полученные из целого цветущего свежего растения по методу 3 "Настойки гомеопатические матричные" Временная фармакопейная статья (ВФС) 42-2799—96 в соотношении сырье—этанол 50% (по объему) 1:3; из высушенных корневищ с корнями по методу 4 "Настойки гомеопатические матричные" ВФС 42-2799—96 в соотношении сырье—этанол 90% (по объему) 1:10. Настойки арники горной представляют собой прозрачные жидкости зеленовато-желтого (из целого цветущего свежего растения) и желто-оранжевого (из высушенных корневищ с корнями) цвета с характерным ароматным запахом. Компонентный состав аминокислот гомеопатических матричных настоек арники горной изучали методом тонкослойной хроматографии (ТСХ), а также с помощью аминокислотного анализатора. В качестве неподвижной фазы использовали пластинки "Сорбфил" (Россия). В качестве подвижных фаз изучали: 1) н-пропанол—вода (70:30); 2) хлороформ—метанол—10% аммиак (40:40:20); 3) н-бутанол—ледяная уксусная кислота—вода (40:10:5); 4) н-бутанол—ледяная уксусная кислота—вода (40:10:10); 5) н-бутанол—ледяная уксусная кислота—вода (40:10:20); 6) н-бутанол—диэтиловый эфир—ледяная уксусная кислота—вода (9:6:3:1). Данные подвижные фазы оценивали по эффективности разделения аминокислот исследуемых настоек. Коэффициент разделения рассчитывали по формуле:  При расчетах мы использовали величины Rf зон, которые можно соотнести с величинами Rf и окраской стандартных образцов веществ сравнения валина и фенилаланина, полученные при хроматографии в указанных подвижных фазах. Используя коэффициент разделения, мы проанализировали все изучаемые подвижные фазы. Коэффициенты разделения для каждой подвижной фазы представлены в табл. 1.  Зоны веществ на хроматограмме обнаруживали 0,5% раствором нингидрина (ГФ XI, вып.2, ст. 124) в ацетоне. Для сравнения использовали 0,1% растворы стандартов аргинина, пролина, аспарагиновой кислоты, серина, аланина, глутами новой кислоты, валина и фенилаланина в 10% водном растворе н-пропанола. Как видно из табл. 1, наиболее оптимальные условия разделения зон адсорбции настоек достигаются при использовании подвижной фазы 4: н-бутанол—ледяная уксусная кислота—вода (40:10:5).  Методика. На стартовую линию хроматографической пластинки наносят по 10 мкл растворов настоек и по 2 мкл растворов свидетелей и хроматографируют восходящим способом на высоту 10 см. Затем хромматограмму высушивают на воздухе при комнатной температуре до удаления запаха растворителей, обрабатывают раствором нингидрина и высушивают в сушильном шкафу при 100—105°С в течение 5—10 мин. Результаты анализа представлены на рисунке и в табл. 2. Как видно из табл. 2, в настойках арники горной обнаружены свободные аминокислоты; в настойке из целого свежего цветущего растения арники горной идентифицированы аргинин, аспарагиновая кислота, серин, аланин, глутаминовая кислота, валин и фенилаланин, в настойке; из высушенных корневищ с корнями арники горной- аргинин, пролин, аспарагиновая кислота, серин, валин, фенилаланин. Идентифицировать все зоны на хроматограммах методом ТСХ затруднительно, так как зоны некоторых аминокислот имеют близкие значения Rf. В связи с этим, чтобы подтвердить данные, полученные методом ТСХ2, и иметь более полные сведения о компонентном составе аминокислот в гомеопатических матричных настойках арники горной из свежесобранного и высушенного сырья, мы применяли аминокислотный анализ. Анализ проводили на анализаторе фирмы "Hitachi" (Япония) модель 835 на стальной колонке (0,4x15 см), заполненной катионообменной смолой марки 2619 (Hitachi Custom Jon—Exchange Resin), в стандартных условиях, обычно используемых для разделения белковых гидролизатов. Разделение аминокислот проводили в 3-буферной системе натрий-цитратных буферных растворов: 0,18 н. — рН 3,25; 0,3 н. — рН 3,9 и 1,6 н. — рН 4,75. Нингидриновый реактив готовили с использование метилового эфира этиленгликоля. Цитратные буферные растворы подавали в колонку по стандартной программе со скоростью 32 мл/ч, нингидриновый реактив — со скоростью 20 мл/ч. После выхода из аналитической колонки разделенные аминокислоты смешивали с нингидриновым реактивом в смесительном блоке в соотношении 2:1 (по объему). Реакция аминокислот с нингидрином проходила за 4 мин при 100°С в реакционной бане. Колориметрическое измерение окрашенных комплексов, образующихся в результате реакции с нингидрином, проводили непрерывно и одновременно при 2 длинах волн. Первичные амины образовывали пурпурное окрашивание, измерение проводили при длине волны 570 нм. Вторичные амины (например, пролин) образовывали желтое окрашивание, измерение проводили при длине волны 440 нм. Оптическая плотность элюата автоматически регистрировалась on-line системой МультиХром для Windows 97 (фирма "Амперсенд", Россия). Площади пиков идентифицированных аминокислот определяли автоматически, а для неидентифицированных соединений регистрировали время выхода и площади пиков. Количество каждой идентифицированной аминокислоты определяли в наномолях и микрограммах в аликвоте, непосредственно использованной для анализа. Затем вычисляли количественное содержание обнаруженных аминокислот в настойках (в мкг/мл), а также процент выхода от общей суммы. Поскольку изучаемые настойки были получены из разных видов сырья (высушенных корневищ с корнями и целого свежего цветущего растения) и при их изготовлении использовали разное соотношение сырья и спирта этилового разной концентрации, пробоподготовка настоек была различной. 1. Настойка из целого свежего цветущего растения. 1 мл настойки выпаривают досуха под вакуумом на роторном испарителе. Сухой остаток растворяют в 300 мкл смеси для гидролиза, состоящей из 12 н. хлористоводородной кислоты и трифторуксусной кислоты в пропорции 2:1 (по объему) с добавлением 0,001% β-меркаптоэтанола. Гидролиз проводят в запаянных стеклянных ампулах при 155°С в течение 1 ч. Затем гидролизат разбавляют водой в 2 раза, выпаривают досуха на роторном испарителе и растворяют в 500 мкл 0,1 н. хлористоводородной кислоты. 75 мкл полученного раствора разводят 0,1 н. хлористоводородной кислотой до 250 мкл. Для анализа используют 145 мкл полученного раствора. 2. Настойка их высушенных корневищ с корнями. 3 мл настойки выпаривают досуха под вакуумом на роторном испарителе. Сухой остаток растворяют в 300 мкл смеси для гидролиза, состоящей из 12 н. хлористоводородной кислоты и трифторуксусной кислоты в пропорции (2:1) (по объему) с добавлением 0,001% β-меркаптоэтанола. Гидролиз проводят в запаянных стеклянных ампулах при 155°С в течение 1 ч. Затем гидролизат разбавляют водой в 2 раза, выпаривают досуха на роторном испарителе и растворяют в 480 мкл 0,1 н. хлористоводородной кислоты. Для анализа используют 145 мкл полученного раствора. Данные о качественном и количественном составе аминокислот в гомеопатических матричных настойках арники горной после гидролиза приведены в табл. 3. Значения даны для α-аминокислот, так как β-аминокислоты не изучали из-за меньших адсорбционных характеристик и, следовательно, затрудений при их регистрировании. Как видно из табл. 3, при определении качественного состава и количественного содержания аминокислот в гомеопатических матричных настойках из целого свежего цветущего растения и высушенных корневищ с корнями арники горной выявлено 20 аминокислот, в том числе 9 незаменимых: треонин, валин, метионин, изолейцин, лейцин, фенилаланин, лизин, гистидин и аргинин. В настойке из целого цветущего свежего растения содержится значительное количество аргинина (33,77%), большое количество глутамина и глутаминовой кислоты (25,23%), а также аспарагина и аспарагиновой кислоты (8,74%), аланина (4,35%), гидроксилизина (3,76%). В настойке из высушенных корневищ с корнями обнаружено большое количество аргинина (32,02%), глутамина и глутаминовой кислоты (21,78%), пролина (20,56%) и гидроксилизина (9,88%).  Выводы 1. Впервые изучен компонентный состав свободных аминокислот гомеопатических матричных настоек арники горной, полученных из высушенных корневищ с корнями и целого свежего цветущего растения арники горной, с помощью метода ТСХ, а также компонентный состав аминокислот после гидролиза с помощью аминокислотного анализатора. 2. В настойках обнаружено 20 аминокислот, в том числе 9 незаменимых. 3. В настойке из целого свежего цветущего растения арники горной методом ТСХ идентифицированы аргинин, аспарагиновая кислота, серин, аланин, глутаминовая кислота, валин, фенилаланин, а в настойке из высушенных корневищ с корнями арники горной аргинин, пролин, аспарагиновая кислота, серин, валин и фенилаланин. 4. С помощью аминокислотного анализатора после кислотного гидролиза в гомеопатических матричных настойках были выявлены дополнительно другие аминокислоты. Настойка из целого цветущего свежего растения содержит в значительном количестве аргинин, глутамин и глутаминовую кислоту, аспарагин и аспарагиновую кислоту, аланин, гидроксилизин. Настойка из высушенных корневищ с корнями содержит большое количество аргинина, глутамина и глутаминовой кислоты, пролина и гидроксилизина. 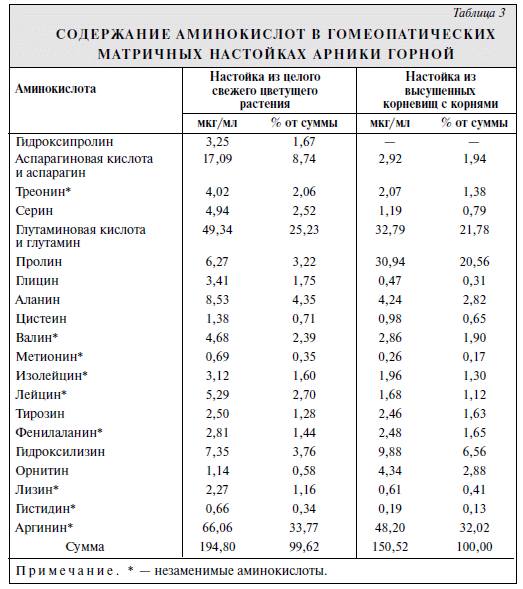
Бициллин-3 является пенициллином пролонгированного действия и представляет собой смесь натриевой, новокаиновой и бензатиновой солей бензилпенициллина (БП). По действующей ВФС 42-3034—98 определение БП в препарате проводят с помощью ВЭЖХ, новокаин определяют спектрофотометрически, а бензатин (N,N1-дибензилэтилендиамин) экстрагируют эфиром из водного раствора, насыщенного хлоридом натрия. После выпаривания эфира бензатин определяют титрованием хлорной кислотой. В Европейской Фармакопее содержание БП и бензатина в бензатиновой соли БП определяют с помощью градиентной ВЭЖХ в смеси метанола с раствором фосфата натрия при рН 3,5. Цель работы — разработка метода ВЭЖХ в изократическом режиме для определения компонентов в бициллине-3. Экспериментальная часть Использовали бициллин-3 производства АКО "Синтез" (Курган). Исследование проводили на хроматографе фирмы "Waters" (США) с насосом модели 510, УФ-детектором модели 481 и инжектором модели 7125 (Rheodyne) с дозирующей петлей вместимостью 50 мкл. Для детектирования использовали длину волны 214 нм, при которой хорошо детектируются все анализируемые соединения. Регистрацию хроматограмм и расчет площадей пиков и основных параметров удерживания проводили с помощью персонального компьютера с аналого-цифровым преобразователем и программой "Мультихром". Был изучен обращеннофазный вариант метода ВЭЖХ на колонке "Luna C18(2)" размером 250 х 4,6 мм фирмы "Phenomenex" (США), поскольку колонка зарекомендовала себя ранее как относительно дешевая с улучшенной симметрией выхода пиков органических аминов. С этой же целью в качестве подвижной фазы использовали смесь ацетонитрила с буферным раствором, содержащим в качестве одного из компонетов триэтиламин, имеющим pH 5,0. Исходный раствор для приготовления подвижной фазы — 2,5 М раствор фосфорной кислоты, который титровали триэтиламином до рН 5,0. Буферный раствор для ВЭЖХ получали разбавлением исходного раствора водой в 10 раз. 750 мл полученного буферного раствора смешивали с 250 мл ацетонитрила. При этом значение кажущегося рН подвижной фазы повышалось до 5,7. Скорость подвижной фазы 1 мл/мин. Хроматографирование проводили при комнатной температуре. Время анализа 20 мин. Поскольку входящие в состав препарата компоненты различаются по кислотно-основным свойствам — БП является кислотой, а новокаин и бензатин — основаниями, при увеличении рН их времена удерживания в интервале рН, где их ионизация меняется, сдвигаются в разные стороны. Поэтому путем изменения рН легко подобрать удобное удерживание анализируемых компонентов. Однако увеличение рН приводит к заметному ухудшению формы пика бензатина, а уменьшение — к недостаточному разрешению новокаина и продуктов гидролиза БП. Разделение компонентов бициллина-3 при указанных выше условиях представлено на рисунке. Времена удерживания новокаина, бензатина и БП составляли 4,2, 11,6 и 14,8 мин соответственно. 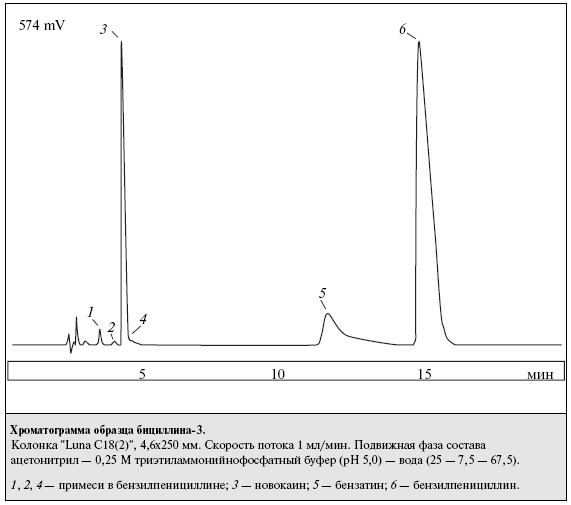 Существенным является выход пика новокаина между 2 пиками, представляющими собой продукты гидролиза БП. В связи с этим для лучшего разделения компонентов рекомендуется добавлять в подвижную фазу небольшие количества 2,5 М растворов фосфорной кислоты или триэтиламина и контролировать разделение хроматографированием смеси новокаина и БП, раствор которого хранился около суток при комнатной температуре. Для количественного определения 20—25 мг бициллина-3 вносили в мерную колбу вместимостью 100 мл и растворяли в 20% водном растворе ацетонитрила. Использование для растворения метанола или его растворов вело к частичному метилированию БП. Увеличение концентрации ацетонитрила приводило к уширению пика новокаина. Верхний предел концентрации лекарственного вещества ограничен его растворимостью. Калибровочные графики для БП и бензатина получали с использованием натриевой соли БП и диацетата бензатина после соответствующего пересчета. Калибровочный график для БП линеен в области 0,1—0,5 мг/мл, для бензатина и новокаина — в области 0,01—0,05 мг/мл. Результаты определения компонентов в 5 сериях препарата представлены в табл. 1, где каждое значение представляет собой среднее из 5 определений. Относительное среднее квадратичное отклонение составило 1,6% для новокаина, 3,4% для бензатина и 1,4% для БП. Из табл. 1 следует, что результаты количественного определения с помощью ВЭЖХ укладываются в допустимые пределы, регламентируемые НД. 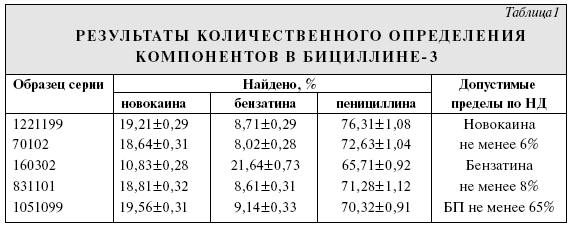 Для подтверждения правильности предложенной методики были проанализированы компоненты бициллина-3 в модельных смесях, приготовленных смешением натриевой соли БП, бензатина диацетата и новокаина. Результаты приведены в табл. 2. Результаты пересчитаны на исходные компоненты. 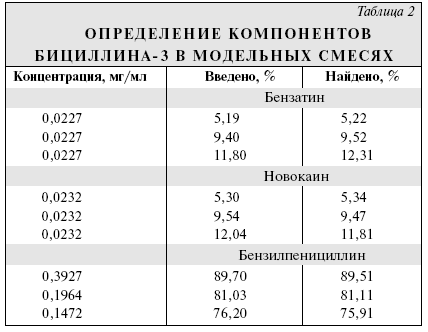 Каждое значение в графе "Найдено" табл. 2 — средний результат 3 определений. Средняя величина относительного отклонения составила 2,2 % для бензатина, 0,9 % для новокаина и 0,8 % для БП, что коррелирует с относительными средними стандартными отклонениями, найденными при анализе компонентов в реальных пробах. Для бензатина разброс результатов несколько выше, чем для остальных компонентов, что объясняется низкой высотой и неправильной формой пика и соответственно большей ошибкой интегрирования. Другой причиной относительно больших ошибок при определении бензатина может явиться эффект памяти инжектора при анализе сильно адсорбирующихся веществ. Однако и такой разброс, несколько превышающий принятый для анализов с помощью метода ВЭЖХ, вполне допустим для определения бензатина. Выводы 1. Разработана методика обнаружения и количественного определения компонентов в препарате "Бициллин-3". 2. Методика проверена на ряде серий препарата и подтверждена анализом модельных смесей известного состава.
Пропифеназон (4-изопропил-2,3-диметил-1-фенил-3-пиразолин-5-он; изопропилантипирин) относится к ненаркотическим анальгетикам пиразолонового ряда и входит в состав комбинированных безрецептурных препаратов. В настоящее время в лечебной практике широко используются таблетки каффетин (состав: пропифеназона—0,21 г, парацетамола—0,25 г, кофеина—0,05 г, кодеина фосфата—0,01 г) и саридон (парацетамола—0,25 г, пропифеназона—0,15 г, кофеина—0,05 г). Согласно существующей нормативной документации для подтверждения подлинности таблеток каффетина используется хроматография в тонком слое сорбента. Количественное определение предложено проводить в отдельных навесках препарата разными для каждого компонента методами: с помощью спектрофотометрии, титриметрии, сочетание тонкослойной хроматографии и спектрофотометрии. В нормативной документации на саридон парацетамол, кофеин и пропифеназон определяют методом ВЭЖХ на колонках длиной 12,5 см с сорбентом Merck Lichrospher C18 и подвижной фазой состава: метанол—0,01 М фосфорная кислота в соотношении 30:70. Цель настоящей работы — разработка методик обнаружения и количественного определения компонентов таблеток каффетина и саридона с помощью ВЭЖХ. В работе использовали таблетки каффетина производства «Алкалоид Скопье» (Республика Македония), саридона производства «Лаборатория Рош Николас С.А.», Гайярд (Франция) и субстанции компонентов, входящих в их состав. Исследование проводили на отечественном микроколоночном жидкостном хроматографе «Милихром-4» с УФ-спектрофотометрическим детектором, колонкой длиной 8 см с обращенно-фазовым сорбентом Сепарон-С18 в качестве неподвижной фазы. Полярный характер анализируемых соединений, их хорошая растворимость в воде и ацетонитриле обусловили выбор водно-ацетонитрильных смесей в разных соотношениях в качестве подвижной фазы. Были испытаны подвижные фазы ацетонитрил — вода в соотношениях 9:1; 7:3; 6:4; 8:2 и ацетонитрил—вода—диэтиламин (3:2:0,2). Избегали использовать подвижные фазы с объемной долей органического растворителя более 80%, чтобы исключить нормально-фазовые взаимодействия, затрудняющие дальнейшее регулирование состава подвижной фазы. Объемная доля растворителя менее 5% приводит к функциональной неустойчивости подвижной фазы и невоспроизводимости времен удерживания . Введение в состав подвижной фазы диэтиламина в качестве модификатора позволило добиться разделения всех 4 компонентов таблеток каффетин. Известно, что на поверхности октадецилсиликагеля находится значительное количество остаточных силанольных групп, способных к ионообменному взаимодействию. Диэтиламин исключает из хроматографического процесса силанольные группы, улучшает форму пиков, сокращает время анализа и регулирует рН на поверхности силикагеля. Измерение проводили в следующих условиях: масштаб регистрации 2,0, время удерживания 0,8 с, скорость расхода элюента 50 мкл/мин, объем вводимой пробы 3 мкл. Детекцию пиков осуществляли при 2 длинах волн — 238 и 276 нм. Идентификацию проводили по параметрам удерживания, которые определяли предварительно на стандартных растворах исследуемых веществ. Компоненты таблеток каффетина разделены при использовании подвижной фазы ацетонитрил—вода—диэтиламин (3:2,2:0,2). Время удерживания парацетамола составило 3,08 мин, пропифеназона — 5,73 мин, кофеина — 4,0 мин, кодеина — 4,67 мин. Компоненты саридона можно также разделить, используя подвижную фазу ацетонитрил—вода (8:2). Время удерживания для парацетамола — 3,9 мин, пропифеназона — 5,11 мин, кофеина — 4,44 мин. Для количественного определения применяли метод абсолютной калибровки. Прямая пропорциональная зависимость концентрации вещества от высоты пика наблюдалась для парацетамола в диапазоне 50—200 мкг/мл, для пропифеназона — 25—128 мкг/мл, кофеина — 20—50 мкг/мл, кодеина — 59—234 мкг/мл. Метод ВЭЖХ имеет некоторые ограничения в анализе сложных смесей. При одновременном присутствии в смеси веществ в макро- и микроколичествах возникает перегрузка колонки, что оказывает влияние на качество разделения и форму выходящих пиков. В каффетине содержание кодеина фосфата по отношению к парацетамолу и пропифеназону в 21—25 раз меньше, поэтому рекомендуется жидкостная экстракция для отделения кодеина от остальных компонентов таблеток. Предварительно мы установили, что парацетамол, пропифеназон и кофеин извлекаются при однократной экстракции этилацетатом из водных растворов при рН 2,0 в количестве соответственно 87,43, 87,29 и 87,84%, а кодеин полностью остается в водном растворе и для его извлечения и концентрирования необходимо использовать хлороформ при рН 9,0—10,0. 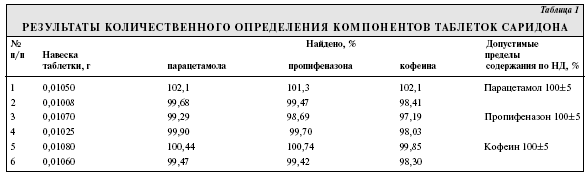 Методика количественного определения парацетамола, пропифеназона и кофеина в таблетках саридона и каффетина. 20 таблеток растирают в ступке в мелкий однородный порошок, отвешивают около 0,01 г (точная навеска) порошка растертых таблеток и помещают в мерную колбу вместимостью 25 мл, прибавляют 10 мл ацетонитрила и тщательно перемешивают. Содержимое колбы доводят до метки ацетонитрилом, перемешивают и фильтруют. Раствор вводят в колонку хроматографа в объеме 3,0 мкл. Содержание парацетамола, пропифеназона и кофеина определяют методом абсолютной калибровки. Результаты определения приведены в табл. 1 и 2, из которых видно, что полученные данные укладываются в допустимые пределы содержания по нормативной документации (НД).  Методика количественного определения кодеина фосфата в таблетках каффетина. Около 0,2 г растертых таблеток (точная навеска) растворяют в 20 мл воды, хорошо перемешивают до получения однородного раствора, фильтруют через фильтр для мелких и самых мелких осадков, фильтр промывают 10 мл очищенной воды. Раствор подкисляют 10% раствором серной кислоты до рН 2,0. Экстрагируют трижды этилацетатом порциями по 10 мл. Экстракты отбрасывают. К водному раствору добавляют 25% раствор аммиака до рН 9,0—10,0. Экстрагируют трижды хлороформом порциями по 10 мл. Объединенные хлороформные вытяжки помещают в фарфоровые чашки и испаряют при комнатной температуре. Сухие остатки растворяют в ацетонитриле, переносят в мерную колбу вместимостью 25 мл и доводят до метки тем же растворителем. 3 мкл полученного раствора вводят в колонку хроматографа и определяют кодеин в описанных условиях. Результаты определения приведены в табл. 3.  Для оценки точности предлагаемых методик и проверки воспроизводимости результатов были приготовлены и исследованы модельные смеси. Данные на примере таблеток саридона приведены в табл. 4. Как видно из табл.4, относительная погрешность определения не превышает для парацетамола ±1,19%, пропифеназона ±1,16%, кофеина ±1,63%. Методика количественного определения компонентов таблеток саридона в модельных смесях. Отвешивают точные навески парацетамола (около 0,08 г), пропифеназона (около 0,05 г) и кофеина (около 0,016 г), переносят в мерную колбу вместимостью 50 мл, растворяют в небольшом объеме ацетонитрила и доводят до метки тем же растворителем. Отбирают аликвоту 2,5 мл и переносят в мерную колбу вместимостью 25 мл, объем до метки доводят тем же растворителем, перемешивают и фильтруют. Раствор вводят в колонку хроматографа в объеме 3 мкл. 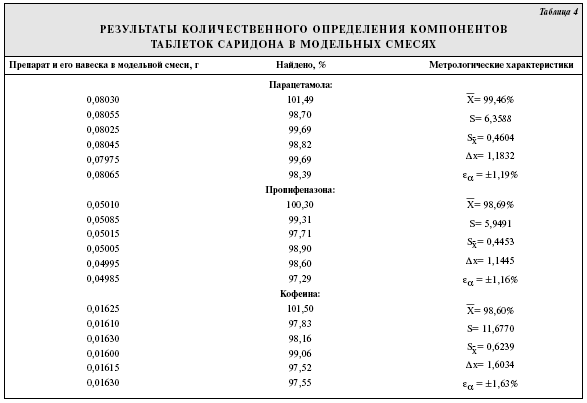 Выводы 1. Разработана методика обнаружения компонентов таблеток каффетина и саридона с помощью ВЭЖХ. Время удерживания для парацетамола составило 3,08 мин, для пропифеназона — 5,73 мин, кофеина — 4 мин и кодеина — 4,67 мин. 2. Предложен метод ВЭЖХ для количественного определения компонентов таблеток каффетина и саридона. Относительная ошибка определения составила для парацетамола ±1,19—1,21%, пропифеназона ±1,16—1,71%, кофеина ±1,22—1,63% и для кодеина ±2,95%.
Цель исследования [2] — определение относительной биодоступности и биоэквивалентности шипучих таблеток «Таспир» производства «Татхимфармпрепараты» (Россия) в сравнении с шипучими таблетками «Upsarin UPSA» фирмы «UPSA» (Франция).Ацетилсалициловая кислота (АСК) широко применяется в медицинской практике в виде шипучих растворимых таблеток, преимуществом которых является уменьшение раздражающего действия кислоты на слизистую желудка. Кроме активного вещества, шипучие таблетки содержат такое соотношение органических пищевых кислот и карбонатов, которое позволяет полностью или частично пройти реакции нейтрализации при попадании таблетки в воду. Казанским производственным химико-фармацевтическим объединением (КПХФО) «Татхимфармпрепараты» разработаны состав и технология растворимых шипучих таблеток «Таспир», содержащих 0,3 г АСК. В состав шипучей смеси введено новое вспомогательное вещество — двухосновная пищевая янтарная кислота, которая входит в число пищевых кислот цикла Кребса, представляет собой универсальный внутриклеточный метаболит, широко участвующий в обменных реакциях в организме человека, стимулирует клеточное дыхание и оказывает кардиопротекторное действие. Установлено усиление противовоспалительной активности АСК в шипучих таблетках в сочетании с янтарной кислотой. Материал и методы. Испытуемый препарат (Т): ≪Таспир≫, таблетки шипучие по 300 мг № 10, КПХФО ≪Татхимфармпрепараты≫ (Россия). Серия препарата — 9032000, срок годности до 04.02. Препарат сравнения (R): ≪Upsarin UPSA≫, таблетки шипучие по 500 мг № 16, фирма ≪UPSA≫ (Франция). Серия — Е0067, срок годности до 01.04. Фармакокинетическое исследование проводили по перекрестной схеме. Для этого 18 здоровых добровольцев — 9 мужчин, средний возраст 26 (21—44) лет, масса тела 71 (62—82) кг, рост 181 (174—194) см и 9 женщин, средний возраст 28 (21—35) лет, масса тела 61 (53—69) кг, рост 169 (164—174) см — методом простой рандомизации разделили на 2 равные группы. В случайном порядке 9 волонтеров принимали сначала испытуемый препарат (таспир), а через 7 дней — препарат сравнения (Upsarin UPSA); добровольцы другой группы принимали препараты в обратном порядке. Препараты принимали per os в 9 ч утра. Мерным цилиндром отмеряли 200 мл кипяченой воды и полностью растворяли в ней 1 таблетку ≪Таспира≫ (300 мг АСК). Затем отмеряли 330 мл кипяченой воды, полностью растворяли в ней 1 таблетку ≪Upsarin UPSA≫ (500 мг) и потом отмеряли 200 мл этого раствора. Добровольцы принимали 200 мл водного раствора одного из препаратов, который содержал 300 мг АСК. Кровь брали из кубитальной вены в количестве 5 мл в стеклянные пробирки до приема препарата и через 10, 15, 20, 30, 45 мин и 1, 1,5, 2, 3, 4, 6, 8 ч после его приема. Пробы крови центрифугировали 15 мин при 4°С и отбирали 1,5 мл плазмы. Анализ проводили не позднее 2 ч после отбора проб. Концентрацию салициловой кислоты в плазме крови добровольцев определяли с помощью высокоэффективной жидкостной хроматографии. Препараты и реактивы: субстанция салициловой кислоты (ФАРМ,≪Авогадро≫, Россия); ацетонитрил (ТУ 6-09-5497—91); диэтиловый эфир (медицинский); о-фосфорная кислота (85%, чда); хлористоводородная кислота (37%); вода — бидистиллированная. Экстракция. К 1 мл плазмы добавляют 200 мкл смеси 0,2 М НСl и 0,2 М о-фосфорной кислоты (50:50), перемешивают на vortex 1—2 с. Затем добавляют 10 мл диэтилового эфира и экстрагируют 10 мин. Пробы центрифугируют 10 мин при 3000 об/мин. Органический слой количественно переносят в колбы для упаривания и упаривают на роторном испарителе досуха. К сухому остатку добавляют 150 мкл подвижной фазы, встряхивают на vortex 10 с и аликвоту (100 мкл) наносят на колонку хроматографа. Анализ проводили на высокоэффективном жидкостном хроматографе ≪Gilson≫ (Франция) с УФ-спектрофотометрическим детектором с переменной длиной волны при длине волны 237 нм. Использовали обращеннофазную хроматографическую колонку: Диасорб 130-С16; 7мкм; 4•150 мм (Россия). Температура колонки 30°С. Элюирование проводили мобильной фазой состава: вода — ацетонитрил — о-фосфорная кислота (74:18:0,09); рН мобильной фазы около 2,5. Мобильную фазу перед использованием дегазировали под вакуумом. Скорость элюирования 1 мл/мин. В указанных условиях время выхода салициловой кислоты на хроматограмме 8,7±0,2 мин. Количественное определение салициловой кислоты проводили методом абсолютной калибровки по площади пиков с использованием интегратора ≪Shimadzu≫. Калибровочная зависимость носила линейный характер в диапазоне концентраций 1—50 мкг/мл. Калибровочный график описывался линейным уравнением вида: у=а+b . х, где у — концентрация препарата (в мкг/мл), а = 0,000027; b = 0,005948; х — площадь хроматографического пика препарата. Коэффициент регрессии r=0,9997. 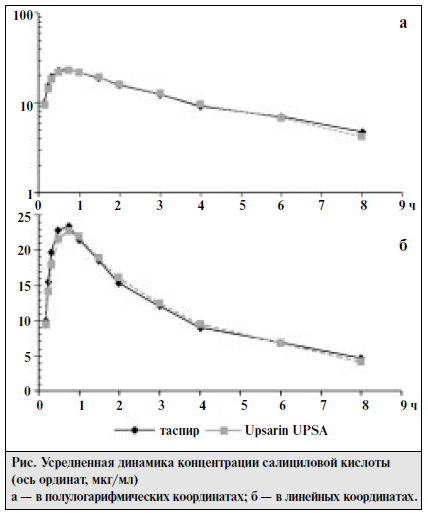 Предел обнаружения 0,5 мкг/мл. Фармакокинетические параметры рассчитывали с помощью программы Kinetica ТМ2000 модельно-независимым методом. Полученные экспериментальные данные статистически обработаны с помощью пакета Systatw5 для персонального компьютера. Результаты исследования Усредненные фармакокинетические кривые салициловой кислоты после однократного перорального приема 300 мг АСК из препаратов ≪Таспир≫ и ≪Upsarin UPSA≫ представлены на рисунке. При анализе усредненных фармакокинетических кривых таспира и Upsarin UPSA видно, что они практически совпадают. После приема как препарата сравнения, так и испытуемого препарата метаболит аспирина салициловая кислота достигал максимального уровня к 45 мин (23,43±1,43 и 22,79±1,55 мкг/мл соответственно). Результаты расчетов фармакокинетических параметров препаратов ≪Таспир≫ и ≪Upsarin UPSA≫ приведены в табл. 1 и 2. Как видно из приведенныхданных, фармакокинетические параметры салициловой кислоты после приема таспира и Upsarin UPSA статистически достоверно не различались. Параметры относительной биодоступности салициловой кислоты после однократного приема шипучих таблетированных лекарственных форм представлены в табл. 3, из которой видно, что среднее значение относительной биодоступности таспира по отношению к Upsarin UPSA составляет 1,02, а величина отношения максимальных концентраций — 1,04. Значения отношений максимальной концентрации к площади под фармакокинетической кривой Cmax/AUCo-∞ для препаратов ≪Таспир≫ и ≪Upsarin UPSA≫ статистически достоверно не различались и составили в среднем 0,22±0,01 и 0,24±0,02 соответственно. Таким образом, не выявлено статистически достоверных различий в процессе всасывания (как по полноте, так и по скорости) препаратов ≪Таспир≫ и ≪Upsarin UPSA≫. Вывод Испытуемый препарат ≪Таспир≫ биоэквивалентен препарату сравнения ≪Upsarin UPSA≫. 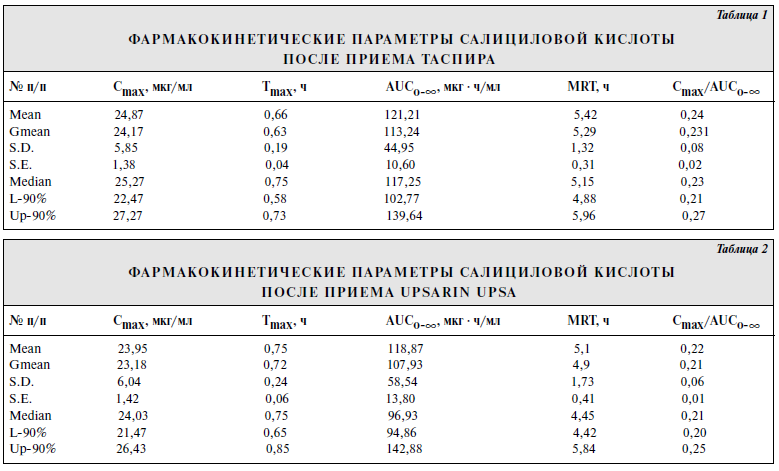 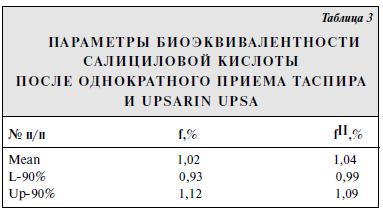
Цель исследования - разработка универсальной методики анализа таблеток "Спазмовералгин нео" сложного состава, содержащих указанные выше ЛВ, которую можно было бы использовать для идентификации, количественного определения, оценки однородности дозирования и высвобождения действующих веществ. В процессе работы исследования влияние различных зна чений рН и компонентного состава подвижной фазы на хроматографичсские характеристики определяемых Л В. Исследования проводили на жидкостном хроматографе LC-10A фирмы "Шимадзу" (Япония). В качестве подвиж ной фазы была применена смесь ацетонитрила с 0,05 М фосфатным буферным раствором в соотношении 20:80. Детектирование проводили с помощью спектрофотометрического детектора при длине волны 230 им. Скорость подвижной фазы 1 мл/мин. Объем вводимой пробы 20 мкт (дозирующая петля). Обработку результатов определения проводили на интеграторе «SP 4100.2 производства фирмы "Спектрафизикс" (США). Результаты оценивали по време нам удерживания и форме хроматографических пиков опре деляемых веществ. На предварительном этапе исследований с целью разра ботки наиболее оптимальной методики, применимой для идентификации и количественной опенки, было проведено исследование влияния состава подвижной фазы на разделе ние изучаемых JIB на хроматографических колонках разме ром 250 х 4,6 мм. заполненных неподвижными фазами раз личной полярности: "Ultrasphere ODS" (5 мкм). "Zorbax С8" (5 мкм), "Ultrasphere CN* (5 мкм). На колонке "Ultrasphere ODS" в данных условиях пол ного разделения хроматографических пиков изучаемых ЛВ добиться не удалось. Кроме того, произошло размывание пика папаверина гидрохлорида вследствие длительного вре мени его удерживания. Добавление ион-парного реагента (гептансульфокислоты) позволило добиться полного разде ления всех компонентов изучаемой смеси, однако увеличи лось время выхода пика папаверина гидрохлорида до 50 мин. С целью уменьшения времени анализа была исследова на возможность применения неподвижной фазы "Zorbax С8" (5 мкм). Полученные результаты показали, что в данных условиях не происходит удовлетворительного разделения компонентов. Наилучшее разделение почти всех Л В было достигнуто при использовании аналитической колонки с сорбентом "Ultrasphere CN" (5 мкм). Однако для разработки методики, пригодной для оценки подлинности и количественного опре деления изучаемых Л В, требовалось провести дальнейшую оптимизацию условий их хроматографического разделения с учетом влияния значения рН подвижной фазы и концентра ции в ней органического компонента. Изучали влияние значения рН подвижной фазы (в пределах от 2,5 до 6,0) на хроматографичсское поведение изучаемых ЛB (рис. I, 2). Для кодеина фосфата, атропина сульфата и в меньшей степени для фенобарбитала наблюда ется закономерность увеличения фактора емкости при воз растании значения рН подвижной фазы. Эффективность колонки для всех исследуемых Л В. кроме папаверина гидро хлорида. в вышеописанных условиях увеличивается при возрастании значения рН. Экспериментально установлено, что оптимальная вели чина рН подвижной фазы находится в пределах от 3,5 до 4.5. На основании этого для дальнейших исследований была ис пользована подвижная фаза со значением рН 4,0. 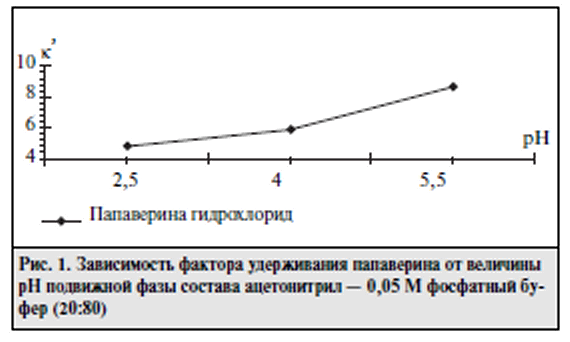 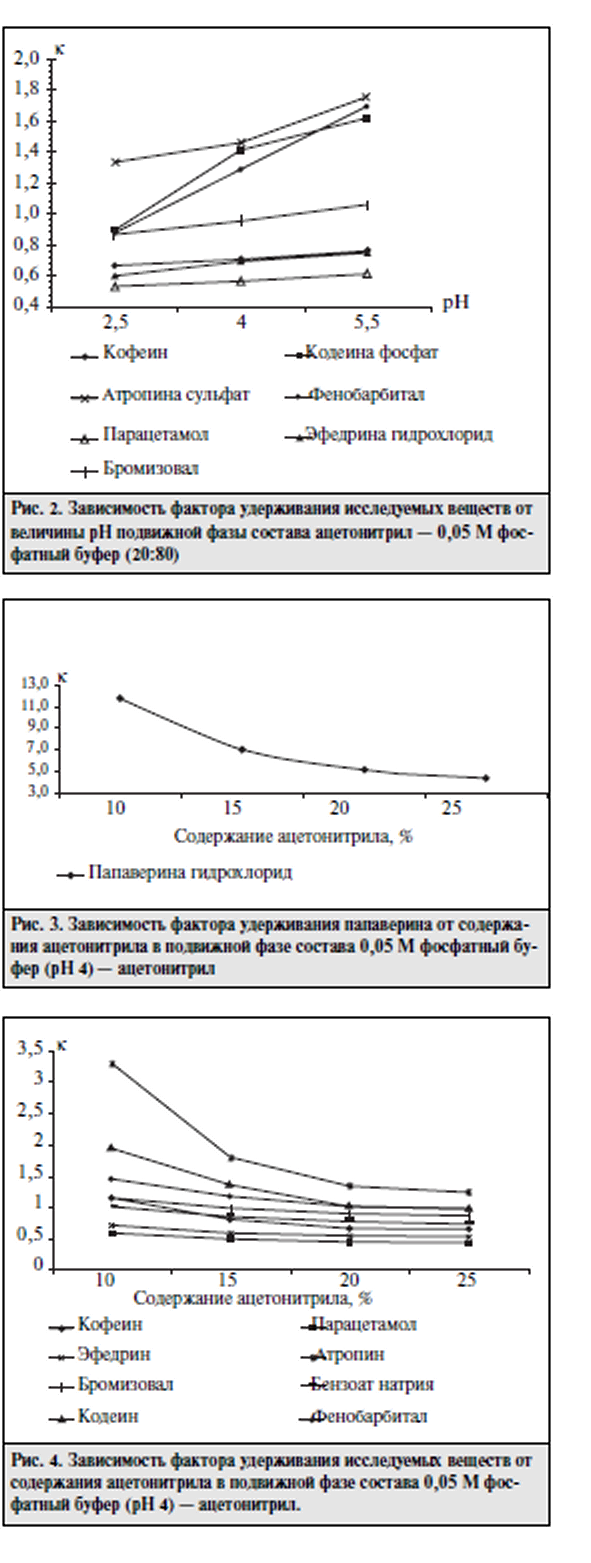 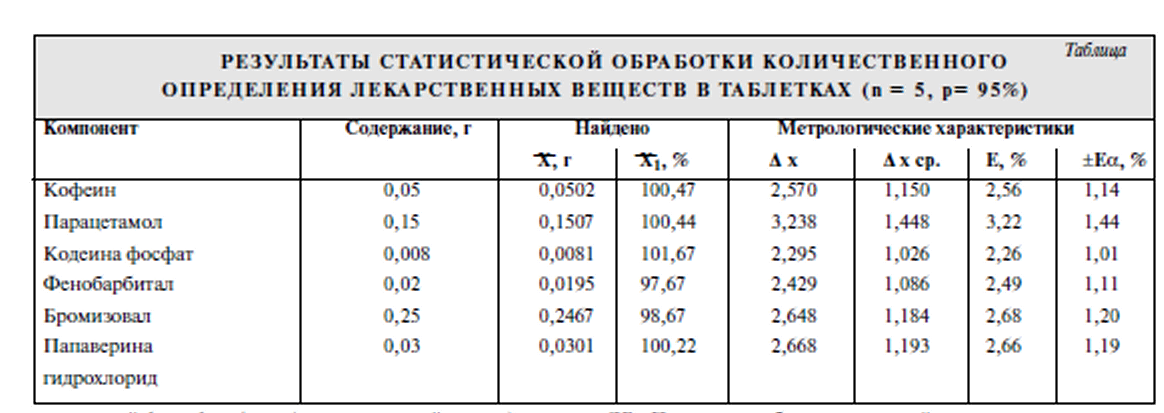 В описанных выше условиях проводился анализ мо дельных смесей исследуемых JIB и полупромышленных об разцов лекарственной формы с целью дальнейшей отработ ки методики идентификации и количественного определе ния. Изучаемые ЛВ в модельных смесях обнаруживаюсь на хроматограмме в следующем порядке: парацетамол, кофе ин, бромизовал, фенобарбитал, кодеина фосфат, атропина сульфат, папаверина гидрохлорид. Однако при выборе ана литических концентраций JIB (в соответствии с их содержа нием в лекарственной форме) и определении пределов их обнаружения выяснилось, что предел обнаружения эфед рина гидрохлорида составляет 1 мкг/мл, атропина сульфата — 0.6 мкг/мл. Концентрации этих ЛВ в растворе для коли чественного определения составляют 1,0 и 0,05 мкг/мл соответственно. Поэтому было предложено определять эти JIB альтернативными методами. Оценку количественного содержания исследуемых ве ществ проводили по методу внешнего стандарта как наибо лее оптимальному для хроматографического анализа смесей 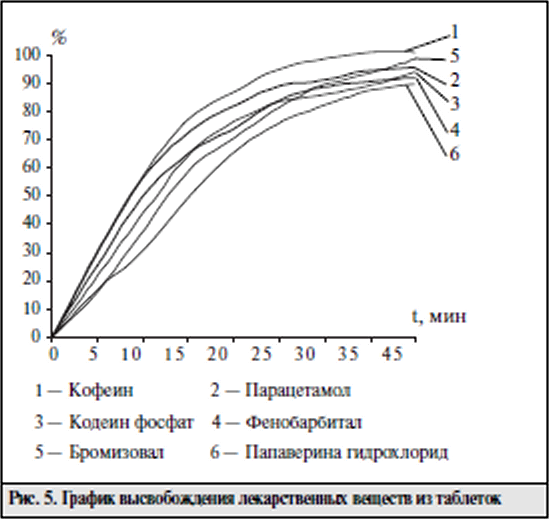 При этом наблюдается линейная зависимость между площадью хроматографического пика и концентрацией определяемого ЛВ в анализируемом рас творе в интервале определяемых концентраций. Для опенки воспроизводимости и точности разработан ной методики проведена серия анализов модельных образцов таблеток. Метрологические характеристики для изучае мых соединении представлены в таблице. Приведенные в таблице данные свидетельствуют о том, что предлагаемая методика с применением ВЭЖХ обладает достаточной точностью и воспроизводимостью и может быть использована для количественного определе ния шести ЛВ в изучаемой таблетированной лекарствен ной форме. В соответствии с требованиями ГФ XI таблетки, со держащие в своем составе ЛВ в количестве 0.05 г и менее, должны тестироваться на однородность дозиро вания. Разработанная методика ВЭЖХ была использова на для определения однородности дозирования четырех компонентов: кофеина, фенобарбитала, кодеина фосфа та и папаверина гидрохлорида. Анализ показал, что по этому показателю таблетки соответствуют требованиям ГФ XI. Выводы: Подобранные хроматографические усло вия использовали для разработки методики теста "Растворе ние" и исследования динамики высвобождения Л В из табле ток. Полученные кривые высвобождения (рис. 5) свидетель ствуют о том, что наибольшая скорость высвобождения на блюдается для кофеина и парацетамола (70% за 15 мин). Од нако и остальные ЛВ в течение 20 мин высвобождаются на 70% и более. Список использованной литературы
|