Определение биологии как науки. Предмет и методы биологии. Человек как объект биологии. Биосоциальная природа человека
 Скачать 2.28 Mb. Скачать 2.28 Mb.
|

|
Изучение ДНК: строение, структура ДНК, функции Для детального понимания сути метода ПЦР-диагностики необходимо совершить небольшой экскурс в школьный курс биологии. Еще из школьных учебников мы знаем, что дезоксирибонуклеиновая кислота (ДНК) — универсальный носитель генетической информации и наследственных признаков у всех существующих на Земле организмов. Исключение составляют только некоторые микроорганизмы, например, вирусы — универсальным носителем генетической информации у них является РНК - одноцепочечная рибонуклеиновая кислота. Строение ДНК-молекулы Открытие ДНК молекулы произошло в 1953 году. Френсис Крик и Джеймс Уотсон открыли структуру двойной спирали ДНК, их работа впоследствии была отмечена Нобелевской премией. ДНК представляет собой двойную нить, скрученную в спираль. Каждая нить состоит из «кирпичиков» — из последовательно соединенных нуклеотидов. Каждый нуклеотид ДНК содержит одно из четырёх азотистых оснований — гуанин (G), аденин (A) (пурины), тимин (T) и цитозин (C) (пиримидины), связанное с дезоксирибозой, к последней, в свою очередь, присоединена фосфатная группа. Между собой соседние нуклеотиды соединены в цепи фосфодиэфирной связью, образованной 3’-гидроксильной (3’-ОН) и 5’-фосфатной группами (5’-РО3). Это свойство обуславливает наличие полярности в ДНК, т. е. противоположной направленности, а именно 5’- и 3’-концов: 5’-концу одной нити соответствует 3’-конец второй нити. Структура ДНК П 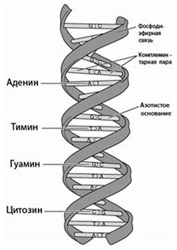 ервичная структура ДНК — это линейная последовательность нуклеотидов ДНК в цепи. Последовательность нуклеотидов в цепи ДНК записывают в виде буквенной формулы ДНК: например — AGTCATGCCAG, запись ведется с 5’- на 3’-конец цепи ДНК. ервичная структура ДНК — это линейная последовательность нуклеотидов ДНК в цепи. Последовательность нуклеотидов в цепи ДНК записывают в виде буквенной формулы ДНК: например — AGTCATGCCAG, запись ведется с 5’- на 3’-конец цепи ДНК.Вторичная структура ДНК образуется за счет взаимодействий нуклеотидов (в большей степени азотистых оснований) между собой, водородных связей. Классический пример вторичной структуры ДНК — двойная спираль ДНК. Двойная спираль ДНК — самая распространенная в природе форма ДНК, состоящая из двух полинуклеотидных цепей ДНК. Построение каждой новой цепи ДНК осуществляется по принципу комплементарности, т. е. каждому азотистому основанию одной цепи ДНК соответствует строго определенное основание другой цепи: в комплемнтарной паре напротив A стоит T, а напротив G располагается C и т.д. Синтез ДНК. Репликация У 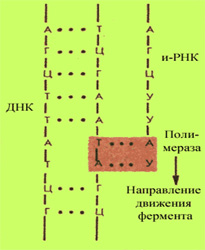 никальным свойством ДНК является ее способность удваиваться (реплицироваться). В природерепликация ДНК происходит следующим образом: с помощью специальных ферментов (гираз), которые служат катализатором (веществами, ускоряющими реакцию), в клетке происходит расплетение спирали в том ее участке, где должна происходить репликация (удвоение ДНК). Далее водородные связи, которые связывают нити, разрываются и нити расходятся. никальным свойством ДНК является ее способность удваиваться (реплицироваться). В природерепликация ДНК происходит следующим образом: с помощью специальных ферментов (гираз), которые служат катализатором (веществами, ускоряющими реакцию), в клетке происходит расплетение спирали в том ее участке, где должна происходить репликация (удвоение ДНК). Далее водородные связи, которые связывают нити, разрываются и нити расходятся.В построении новой цепи активным «строителем» выступает специальный фермент — ДНК-полимераза. Для удвоения ДНК необходим также стратовый блок или «фундамент», в качестве которого выступает небольшой двухцепочечный фрагмент ДНК. Этот стартовый блок, а точнее - комплементарный участок цепи родительской ДНК — взаимодействует с праймером — одноцепочечным фрагментом из 20—30 нуклеотидов. Происходит репликация или клонирование ДНК одновременно на обеих нитях. Из одной молекулы ДНК образуются две молекулы ДНК, в которых одна нить от материнской молекулы ДНК, а вторая, дочерняя, вновь синтезированная. Таким образом, процесс репликации ДНК (удваивания) включает в себя три основных этапа:
В основе анализа методом ПЦР лежит принцип репликации ДНК — синтеза ДНК, который современным ученым удалось воссоздать искусственно: в лаборатории врачи вызывают удвоение ДНК, но только не всей цепи ДНК, а ее небольшого фрагмента. Функции ДНК Молекула ДНК человека — носитель генетической информации, которая записана в виде последовательности нуклеотидов с помощью генетического кода. В результате описанной выше репликации ДНК происходит передача генов ДНК от поколения к поколению. Изменение последовательности нуклеотидов в ДНК (мутации) может приводить к генетическим нарушениям в организме. Билет 29 Моногенные, хромосомные и мультифакториальные болезни человека, механизмы их возникновения и проявления. Примеры. • моногенные болезни Моногенным называется такой тип наследования, когда наследственный признак контролируется одним геном. Моногенные заболевания подразделяются по типу наследования: аутосомно-доминантные (то есть, если хоть один из родителей болен, то и ребенок будет болеть), например -синдром Марфана, нейрофиброма-тоз, ахондроплазия – аутосомно-рецессивные (ребенок может заболеть, если оба родителя носители этого заболевания, или один родитель болен, а второй – носитель мутаций гена, вызывающих это заболевание) – муковисцидоз, спинальная миоатрофия. Пристальное внимание к этой группе болезней обусловлено и тем, что, как оказалось, число их значительно выше, чем думали раньше. У всех болезней совершенно различная распространенность, которая может колебаться в зависимости и от географии и от национальности, например, хорея Хангтингтона встречается у 1 на 20 000 европейцев и почти не встречается в Японии, болезнь Тея-Сакса характерна для евреев-ашкенази и крайне редка у других народов. В России наиболее распространнеными моногенно наследуемыми заболеваниями являются муковисцидоз (1/12000 новорожденных), группа миоатрофий (1/10000 новорожденных), гемофилия А (1/5000 новорожденных мальчиков). Конечно, многие моногенные заболевания выявлены уже давно и хоро¬шо известны медицинским генетикам. К хромосомным относятся болезни, обусловленные геномными мутациями или структурными изменениями отдельных хромосом. Хромосомные болезни возникают в результате мутаций в половых клетках одного из родителей. Из поколения в поколение передаются не более 3—5 % из них. Хромосомными нарушениями обусловлены примерно 50 % спонтанных абортов и 7 % всех мёртворождений. Все хромосомные болезни принято делить на две группы: аномалии числа хромосом и нарушения структуры хромосом. Аномалии числа хромосом Болезни, обусловленные нарушением числа аутосом (неполовых) хромосом синдром Дауна — трисомия по 21 хромосоме, к признакам относятся: слабоумие, задержка роста, характерная внешность, изменения дерматоглифики; синдром Патау — трисомия по 13 хромосоме, характеризуется множественными пороками развития, идиотией, часто — полидактилия, нарушения строения половых органов, глухота; практически все больные не доживают до одного года; синдром Эдвардса — трисомия по 18 хромосоме, нижняя челюсть и ротовое отверстие маленькие, глазные щели узкие и короткие, ушные раковины деформированы; 60% детей умирают в возрасте до 3-х месяцев, до года доживают лишь 10%, основной причиной служит остановка дыхания и нарушение работы сердца. Болезни, связанные с нарушением числа половых хромосом Синдром Шерешевского — Тёрнера — отсутствие одной Х-хромосомы у женщин (45 ХО) вследствие нарушения расхождения половых хромосом; к признакам относится низкорослость, половой инфантилизм и бесплодие, различные соматические нарушения (микрогнатия, короткая шея и др.); полисомия по Х-хромосоме — включает трисомию (кариотии 47, XXX), тетрасомию (48, ХХХХ), пентасомию (49, ХХХХХ), отмечается незначительное снижение интеллекта, повышенная вероятность развития психозов и шизофрении с неблагоприятным типом течения; полисомия по Y-хромосоме — как и полисомия по X-хромосоме, включает трисомию (кариотии 47, XYY), тетрасомию (48, ХYYY), пентасомию (49, ХYYYY), клинические проявления также схожи с полисомией X-хромосомы; Синдром Клайнфельтера — полисомия по X- и Y-хромосомам у мальчиков (47, XXY; 48, XXYY и др.), признаки: евнухоидный тип сложения, гинекомастия, слабый рост волос на лице, в подмышечных впадинах и на лобке, половой инфантилизм, бесплодие; умственное развитие отстает, однако иногда интеллект нормальный. Болезни, причиной которых является полиплоидия триплоидии, тетраплоидии и т. д.; причина — нарушение процесса мейоза вследствие мутации, в результате чего дочерняя половая клетка получает вместо гаплоидного (23) диплоидный (46) набор хромосом, то есть 69 хромосом (у мужчин кариотип 69, XYY, у женщин — 69, XXX); почти всегда летальны до рождения Мультифакториальные заболевания, или болезни с наследственным предрасположенность Группа болезней отличается от генных болезней тем, что для своего проявления нуждается в действии факторов внешней среды. Среди них также различают моногенные, при которых наследственная предрасположенность обусловлена одним патологически измененным геном, и полигенные. Последние определяются многими генами, которые в нормальном состоянии, но при определенном взаимодействии между собой и с факторами среды создают предрасположение к появлению заболевания. Они называются мультифакториальными заболеваниями (МФЗ). Заболевания моногенные с наследственным предрасположением относительно немногочисленны. К ним применим метод менделевского генетического анализа. Учитывая важную роль среды в их проявлении, они рассматриваются как наследственно обусловленные патологические реакции на действие различных внешних факторов (лекарственных препаратов, пищевых добавок, физических и биологических агентов), в основе которых лежит наследственная недостаточность некоторых ферментов. К таким реакциям могут быть отнесены наследственно обусловленная непереносимость сульфаниламидных препаратов, проявляющаяся в гемолизе эритроцитов, повышении температуры при применении общих анестезирующих средств. У человека описана мутация, обусловливающая патологическую реакцию на загрязнение атмосферы, которая проявляется в раннем развитии эмфиземы легких (в возрасте 30—40 лет). У генетически чувствительных индивидов нежелательные реакции могут вызывать некоторые компоненты пищи и пищевые добавки. Известна непереносимость у ряда людей молочного сахара —лактозы. Гены непереносимости лактозы широко распространены среди азиатского населения (до 95—100%) и среди американских негров и индейцев (до 70—75%). У некоторых людей наблюдается непереносимость к употребляемым в пищу конским бобам, вызывающим у них гемолиз. Ряд лиц не переносит жирной пищи и в раннем возрасте страдает атеросклерозом, что повышает риск инфаркта миокарда. У некоторых людей употребление в пищу сыра и шоколада провоцирует мигрень. Отмечены специфические реакции людей на алкоголь. Консерванты и пищевые красители у некоторых людей не подвергаются нормальному усвоению, что также проявляется в непереносимости этих компонентов пищи. Билет 30 Понятие о болезнях с нетрадиционным наследованием (митохондриальные, болезни импритинга, болезни экспансии тринуклеотидных повторов). Примеры. Общие подходы к лечению наследственных болезней. Митохондриальные болезни. Начиная с конца 80-х годов XX века получены убедительные доказательства связи некоторых видов наследственной патологии у человека с мутациями митохондриальной ДНК (см. гл. 4.1) В зависимости от типа мутаций митохондриальные болезни разделяют на 4 группы: а) болезни, вызванные точковыми мутациями, приводящими к замене консервативных аминокислот в собственных белках митохондрий. К ним относятся пигментный ретинит и нейроофтальмопатияЛебера, при которой наступает двусторонняя потеря зрения. Выраженность клинических признаков у больных этими заболеваниями коррелирует с количеством мутантной мтДНК, которое у разных больных может варьировать от 5 до 100% всей мтДНК; б) болезни, вызванные мутациями в генах т-РНК, приводящими к многочисленным дегенеративным заболеваниям с различной степенью тяжести клинических проявлений, коррелирующей с количеством мутантной мтДНК; в) болезни вызванные делениями и дупликациями участков митохондриалъных генов. У человека описано тяжелое заболевание молодого и среднего возраста — отсроченнаякардиопатия, при которой обнаружены делециимтДНКкардиоцитов. Заболевание носит семейный характер. В ряде случаев предполагается Х-сцепленное наследование, что позволяет думать о существовании ядерного гена, мутация которого вызывает делению до 50% мтДНКкардиоцитов; г) болезни, вызванные снижением числа копий мтДНК, что является следствием определенных мутаций. К данной группе относятся летальная инфантильная дыхательная недостаточность и синдром молочнокислого ацидоза, при которых число копий мтДНК снижается до 1—2% от нормы. Снижение содержания мтДНК в клетках различных органов приводит к развитию миопатий, нефропатий, печеночной недостаточности и т.д. вследствие ослабления синтеза белков, кодируемых мтДНК. Изменения в ДНК митохондрий сопровождаются нарушением их функций, связанных с клеточным дыханием. Это определяет характер и степень тяжести клинических проявлений митохондриалъных болезней. Выдвинута также гипотеза о том, что накопление спонтанно возникающих мутаций мтДНК является звеном механизмов старения и развития дегенеративных процессов у человека. Болезни экспансиитринуклеотвдных повторов с явлением антиципации. Под генетической антиципацией (или упреждением) понимается более раннее проявление и возрастание тяжести симптомов наследственного заболевания в последующих поколениях родословной. Антиципация реально проявляется при определенных видах моногенной неврологической патологии, а также при некоторых мультифакториальных заболеваниях. Феномен экспансии числа тринуклеотидных повторов был впервые обнаружен при исследовании синдрома Мартина—Белла или синдрома фрагильной (ломкой) Х-хромосомы, основным фенотипическим проявлением которого является умственная отсталость. Синдром ломкой Х-хромосомы характеризуется довольно широкой распространенностью в популяции (1:1000) и необычным характером наследования. Лишь у 80% мужчин-носителей мутантного локуса имеются клинические и цитогенетические признаки заболевания. 20% носителей как клинически, так и цитогенетически нормальны, но после передачи мутации всем своим дочерям они могут иметь пораженных внуков. Неэкспрессируемый мутантный ген в таком случае становится экс-прессируемым в последующих поколениях. Таким образом мутантный ген при синдроме ломкой Х-хромосомы может существовать в двух формах, отличающихся по своей пенетрантности. Одна — фенотипически не проявляющаяся — премутация, которая при прохождении через женский мейоз превращается в другую форму — полную мутацию. При таком необычном способе наследования и фенотипического проявления мутантного гена, отличном от классического Х-сцепленного наследования, обнаруживается феномен антиципации — более тяжелое проявление заболевания в последующих поколениях. В основе клинических проявлений и цитологической нестабильности в локусе, ответственном за синдром ломкой Х-хромосомы, лежит многократное увеличение повторов тринуклеотида ЦГГ. В норме число повторов колеблется от 5 до 50. Премутация — неэкспрессируемая форма — характеризуется увеличением числа повторов до 50—200. Возрастание числа повторов тринуклеотида ЦГГ свыше 200 приводит к клинической манифестации заболевания и цитогенетическому проявлению ломкой Х-хромосомы. Как правило, у пораженных лиц наблюдается также аномальное метилирование ДНК, приводящее к репрессированию гена. Интересно, что переход от состояния премутации к полной мутации возникает при передаче от матери, причем экспансия ЦГГ-повторов значительно выше при передаче от матери к сыну, чем от матери к дочери. Антиципация, характерная для синдрома ломкой Х-хромосомы, объясняется четкой связью между числом тринуклеотидных повторов и тяжестью клинических проявлений заболевания с цитологической экспрессией ломкости Х-хромосомы. Увеличение числа тринуклеотидных повторов и связанное с этим явление антиципации обнаружены при целом ряде заболеваний (табл. 6.3). Например, при аутосомно-доминантном заболевании—хорее Гетинггона выявляется четкая корреляция между числом ЦАГ-повторов и возрастом дебюта заболевания. У потомков пораженных отцов обнаруживается более тяжелое клиническое течение заболевания. Экспансия числа тринуклеотидных повторов происходит в мужском гаметогенезе. Болезни импринтинга. Особенности наследования и фенотипического проявления при болезнях импринтинга обусловлены явлением геномного импринтинга (ГИ) (импринтинг от англ. imprinting — запечатление). Явление геномного импринтинга связывают со специфическими изменениями хромосом или их участков во время образования мужских и женских гамет. Этим объясняется дифференциальная маркировка отцовских и материнских хромосом у потомков. Точные механизмы дифференциальной маркировки хромосом или их участков в сперматогенезе или овогенезе пока окончательно не выяснены. Однако, немаловажная роль, вероятно, принадлежит процессам специфического метилированияцитозиновых оснований ДНК, выключающим транскрипцию гена. Импринтированные участки в хромосомах определенного родительского происхождения (отцовских иди материнских) избирательно репрессируются у потомка. В связи с этим фенотипически проявляется только информация, полученная от другого родителя, т.е. имеет место моноаллельная экспрессия. Следовательно, фенотипическое проявление мутантного аллеля зависит от того с какой половой клеткой (яйцеклеткой или сперматозоидом) он был передан потомку. Связь этиологии ряда наследственных заболеваний с феноменом ГИ может быть прослежена на разных уровнях организации генетического материала. На геномном уровне организации наследственного материала доказательством роли ГИ в патологии служит различное фенотипическое проявление триплоидных состояний при разном соотношений гаплоидных наборов отцовского и материнского происхождения. У диандрическихтриплоидов (соотношение числа гаплоидных наборов отца и матери 2:1) и у дигеническихтриплоидов (соотношение 1:2) патологические отклонения в развитии плаценты и собственно зародышевых тканей проявляются по-разному. Это свидетельствует о неравноценности функционирования гаплоидных наборов отца и матери в тканях зародыша и плаценты Связь феномена ГИ с патологией на уровне отдельных хромосом можно проследить в случае однородительскойдисомии (ОРД), при которой происходит удвоение хромосомы одного из родителей при утрате гомологичной хромосомы другого родителя. В основе возникновения ОРД лежит нарушение процессов гаметогенеза. При нерасхождении сестринских хроматид в анафазе II мейоза появляются гаметы, в галлоидном наборе которых присутствуют две генетически идентичные хромосомы (изодисомия). В случае нерасхождения гомологичных хромосом в анафазе I мейоза образуются гаметы, в гаплоидном наборе которых имеется пара гомологичных, генетически неидентичных хромосом (гетеродисомия). В обоих случаях гаметы данного индивида дисомны по одной из хромосом. При оплодотворении дисомных гамет нулисомными по той же хромосоме подовыми клетками происходит комплемеитация гамет, приводящая к возникновению нормального диплоидного кариотипа зиготы. Однако в генотипе такой зиготы присутствует двойной набор генов данной хромосомы, происходящих от одного, а не от обоих родителей. Иногда оплодотворение дисомных гамет нормальными половыми клетками сопровождается «коррекцией трисомии» в результате потери сверхчисленной хромосомы. Если при этом сохраняются две хромосомы, пришедшие от одного родителя, то наблюдается явление ОРД. Наконец, состояние ОРД по отдельным локусам хромосом может возникать в результате соматической рекомбинации — кроссинговера между хроматидами гомологичных хромосом, происходящего в соматических клетках (см. рис 3.73). Когда хромосома не содержит импринтированных участков, при ОРД по данной хромосоме может не наблюдаться аномалий фенотипа. Исключением может быть проявление аутосомно-рецессивного заболевания как результат гомозиготизации по рецессивномуаллелю при изодисомии. Если хромосома содержит импринтированные участки, то при возникновении однородительскойдисомии локализованные в них аллели могут быть либо экспрессированы, либо инактивированы в зависимости от родительского происхождения ОРД. Это может стать причиной возникновения патологических отклонений в развитии организма. Фенотипическое проявление при ОРДмат и ОРДотц может быть сходным или прямо противоположным. Возможен летальный эффект уже на ранних сроках развития. Из сказанного выше следует, что в проксимальном районе длинного плеча 15-й хромосомы имеются близкорасположенные и противоположно импринтированные локусы, отвечающие за возникновение фенотипически различных синдромов Прадера — Вилли и Энгельмана. Таким образомимпринтироваться могут участки хромосом разного родительского происхождения, что и определяет нетрадиционное наследование многих патологических состояний, обусловленных мутациями локусов, подверженных импринтингу. |