Основы химической термодинамики
 Скачать 2.79 Mb. Скачать 2.79 Mb.
|

Основное уравнение термодинамики. Это уравнение объединяет формулы первого и второго начала термодинамики:Q = dU + р dV, (3.42) ^Вместо Q в уравнение, выражающее второе начало термодинамики, подставим его выражение (3.42): В заключение еще раз отметим, что если первый закон термодинамики содержит энергетический баланс процесса, то второй закон показывает его возможное направление.

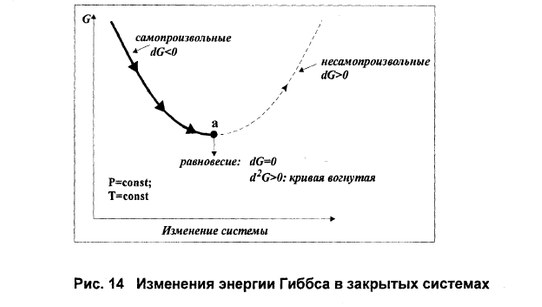
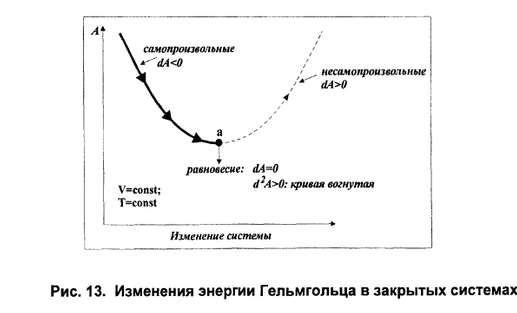
Если процесс протекает самопроизвольно, то внутренняя энергия (энтальпия) должны уменьшаться, а энтропия увеличиваться. Для сравнения этих величин их надо выразить в одних единицах, а для этогоΔS умножить на T. В этом случае имеем ΔН – энтальпийный фактор и ТΔS- энтропийный фактор. В ходе реакции частицы стремятся к объединению, что ведет к уменьшению энтальпии (ΔН < 0), с другой стороны – должна возрастать энтропия, т.е. увеличиваться число частиц в системе (ТΔS > 0). "Движущая сила" реакции определяется разностью между этими величинами и обозначается ΔG. ΔGp,T = ΔH – TΔS и называется изменением энергии Гиббса (изобарно-изотермический потенциал). Энергия Гиббса - это часть энергетического эффекта реакции, которую можно превратить в работу, поэтому ее называют свободной энергией. Это тоже термодинамическая функция состояния и, следовательно, для реакции bB + dD =lL + mM, энергию Гиббса химической реакции можно рассчитать как сумму энергий Гиббса образования продуктов реакции за вычетом энергий Гиббса образования исходных веществ с учетом стехиометрических коэффициентов по формуле: ΔG = lΔfGL + mΔfGM – dΔfGD – bΔfGB. где ΔfG –энергия Гиббса образования веществ. Энергия Гиббса образования веществ это изменение энергии Гиббса системы при образовании 1 моль вещества из простых веществ, устойчивых при 298 К. Энергия Гиббса образования простых веществ ΔfG принимается равной нулю. Если образующееся вещество и исходные простые вещества находятся в стандартных состояниях, то энергия Гиббса образования называется стандартной энергией Гиббса образования вещества ΔfG0. Ее значения приводятся в справочниках. Полученное значение ΔG является критерием самопроизвольного течения реакции в прямом направлении, если ΔG< 0. Химическая реакция не может протекать самопроизвольно в прямом направлении, если энергия Гиббса системы возрастает, т.е. ΔG > 0. Если ΔG = 0, то реакция может протекать как в прямом, так и в обратном направлениях, т.е. реакция обратима. Направление химических реакций зависит от их характера. Так, условие ΔG < 0 соблюдается при любой температуре для экзотермических реакций (ΔН < 0), у которых в ходе реакции возрастает число молей газообразных веществ, и, следовательно, энтропия (ΔS > 0). У таких реакций обе движущие силы (ΔН) и (ТΔS) направлены в сторону протекания прямой реакции и ΔG < 0 при любых температурах. Такие реакции являются необратимыми. Наоборот, эндотермическая реакция (ΔН > 0), в результате которой уменьшается число молей газообразных веществ (ΔS< 0) не могут протекать самопроизвольно в прямом направлении при любой температуре, т.к. всегда ΔG > 0. Если в результате экзотермической реакции (ΔН < 0) уменьшается число молей газообразных веществ и, соответственно, энтропия (ΔS < 0), то при невысокой температуре ΔН > TΔS и реакция возможна в прямом направлении (ΔG < 0). При высоких температурах ΔH < TΔS и прямая реакция самопроизвольно протекать не может (ΔG > 0), а обратная реакция возможна. Для определения температуры равновесия можно воспользоваться условием: Тр = ΔН/ΔS, где Тр – температура, при которой устанавливается равновесие, т.е. возможность протекания прямой и обратной реакций. Если в результате эндотермической реакции (ΔН > 0) увеличивается число молей газообразных веществ и энтропия системы (ΔS > 0), то при невысоких температурах, когда ΔН > ТΔS, самопроизвольно прямая реакция идти не может (ΔG> 0), а при высоких температурах, когда ΔН < TΔS , прямая реакция может протекать самопроизвольно (ΔG < 0). Связь между ΔG и ΔG0 выражается уравнением изотермы Вант-Гоффа, которая для реакции bB + dD = lL + mM записывается в виде: где - относительные парциальные давления соответствующих веществ; концентрации соответствующих растворенных веществ. Итак, энергия Гиббса позволяет определить возможность протекания реакции расчетным путем, не прибегая к дорогостоящим и длительным экспериментам. В изохорно-изотермических условиях свободная энергия называется энергией Гельмгольца или изохорно-изотермическим потенциалом и равна Она характеризует направление и предел самопроизвольного течения химической реакции при изохорно-изотермических условиях, которое возможно при ΔF < 0
Зависимость энергии Гиббса от температуры и давления H – TS U + pV– TS G (7.21) GpTS, H -T rGpT rH -TrS(7.22) H характеризует общее изменение энергии системы в ходе процесса, а изменение энергии Гиббса показывает, какая часть ее может быть превращена в полезную работу, поэтому раньше ее называли свободной энергией, в противоположность энтропийному члену, который характеризовал связанную энергию, недоступную для превращения в работу. а) Из уравнения 7.20а следует, что с ростом давления энергия Гиббса увеличивается, так как V> 0, но темп роста замедляется, так как объем постепенно уменьшается. Причем для разных идеальных газов эти кривые будут одинаковыми. 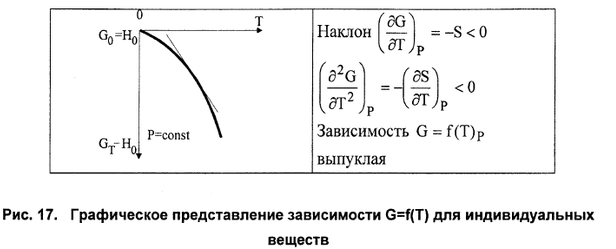  Рис. 7.1. Зависимость энергии Гиббса от давления и температуры. С ростом температуры энергия Гиббса убывает, так как энтропия всегда положительна, причем падение энергии Гиббса будет ускоряться из-за роста энтропии при повышении температуры.    ГИББСА - ГЕЛЬМГОЛЬЦА УРАВНЕНИЯ - термо-динамич. соотношения, устанавливающие связь между внутренней энергией и Гелъмголъца энергией (свободной энергией) F или между энтальпией H и Гиббса энергией (свободной энтальпией) G: Макс. работа, к-рую может совершить система в тепловом контакте с окружающей средой Эта макс. работа за вычетом работы против сил давления P(V2- V1)(максимальная полезная работа)
Раствором называется гомогенная, молекулярно-дисперсная система, состав которой можно изменять непрерывно в некотором конечном или бесконечном интервале. По агрегатному состоянию растворы разделяются на твердые, жидкие и газообразные. Если растворитель и растворенное вещество имеют разные агрегатные состояния, то растворителем рассматривают обычно то вещество, агрегатное состояние которого совпадает с агрегатным состоянием раствора. Если же компоненты раствора и раствор имеют одинаковое агрегатное состояние, то за растворитель считают то вещество, которого больше, хотя для термодинамики это безразлично. Состав раствора измеряется его концентрацией. Существуют следующие основные определения концентрации: мольная доля (х) – число молей вещества в 1 моле раствора; моляльность (m) – число молей растворенного вещества в 1000 г растворителя; молярность (с) – число молей растворенного вещества в 1 л раствора; массовое содержание (р) – число грамм растворенного вещества в 100 г раствора. Характеристические функции многокомпонентных систем. Первый и второй законы термодинамики, из которых следуют фундаментальные уравнения, были получены для закрытых систем, т.е. систем, процессы в которых не приводят к изменению количества компонентов. Реально же чаще встречаются системы, в которых при различных процессах изменяются количества компонентов. Это может происходить, скажем, при фазовых превращениях или вследствие протекания химической реакции. При этом может изменяться состав, как отдельных частей, так и системы в целом. Поэтому внутренняя энергия (и другие функции состояния) открытых систем будут изменяться не только за счет сообщения системе теплоты и произведенной системой работы, но и за счет изменения состава системы. Следовательно для открытых систем характеристические функции будут функциями не только их двух естественных переменных, но и функциями числа молей всех веществ , составляющих систему: U = U ( S, v, n1……………….nk ), H = H ( S, p, n1 ………….. nk ), F = F ( T, v, n1…………… nk ), G = G ( T, p, n1……………nk ). Хими́ческий потенциа́л — термодинамическая функция, применяемая при описании состояния систем с переменным числом частиц. Определяет изменение термодинамических потенциалов (энергии Гиббса, внутренней энергии, энтальпии и т. д.) при изменении числа частиц в системе. Представляет собой энергиюдобавления одной частицы в систему без совершения работы. Определение химического потенциала можно записать в виде: Эта формула определяет, кроме химического потенциала , также давление P и температуру T. Для систем, состоящих из одного компонента, можно доказать, что химический потенциал задаётся формулой Если энергия системы зависит не от объёма, а от других термодинамических параметров , исходная формула принимает вид Если в системе имеется несколько разных типов частиц, есть столько же разных химических потенциалов. Связь химического потенциала со свободной энергией называется Большим термодинамическим потенциалом. Условия равновесия в многокомпонентных гетерогенных системах. Пусть р, Т =Const, тогда . Если в системе имеются фазы (') и (''), то при переходе dni молей i–ого компонента из фазы (') в фазу ('') изменение энергии Гиббса системы dGбудет складываться из изменении энергий Гиббса обеих фаз dG'иdG'': . Если система закрытая, то , а если она к тому же и равновесна, то dG= 0 и, следовательно, , т.е. химические потенциалы данного компонента во всех фазах системы, находящейся в равновесии, равны между собой. Самопроизвольный неравновесный процесс может идти только в сторону уменьшения энергии Гиббса системы dG< 0 (p,T=const). Предположим, что , т.е. фаза (') теряет i–ый компонент, но , значит . Если же , тогда . Таким образом, компонент самопроизвольно переходит из фазы, в которой его химический потенциал больше, в фазу, в которой его химический потенциал меньше. Эти условия равновесия и самопроизвольности процессов переноса вещества в многокомпонентных системах являются обобщением таковых для однокомпонентных систем (см. гл. 6). Кроме того, легко заметить, что химический потенциал чистого вещества равен его мольной энергии Гиббса. ХИМИЧЕСКОЕ РАВНОВЕСИЕ
Химическое равновесие — состояние химической системы, в котором обратимо протекает одна или несколько химических реакций, причём скорости в каждой паре прямая-обратная реакция равны между собой. Для системы, находящейся в химическом равновесии, концентрации реагентов, температура и другие параметры системы не изменяются со временем А2 + В2 ⇄ 2AB В химической термодинамике закон действующих масс связывает между собой равновесные активности исходных веществ и продуктов реакции, согласно соотношению: — активность веществ. Вместо активности могут быть использованы концентрация (для реакции в идеальном растворе), парциальные давления (реакция в смеси идеальных газов), фугитивность (реакция в смеси реальных газов); — стехиометрический коэффициент (для исходных веществ принимается отрицательным, для продуктов — положительным); — константа химического равновесия. Индекс «a» здесь означает использование величины активности в формуле. На практике в расчётах, не требующих особой точности, значения активности обычно заменяются на соответствующие значения концентраций (для реакций в растворах) либо парциальных давлений (для реакций между газами). Константу равновесия при этом обозначают или соответственно. Впервые закон действующих масс был выведен из кинетических представлений Гульдбергом и Вааге, а термодинамический вывод его дан Вант-Гоффом в 1885 году[3]. Пример: для стандартной реакции константа химического равновесия определяется по формуле При наступлении химического равновесия в системе, когда скорость прямой реакции равна скорости обратной реакции и закон действующих масс формулируется следующим образом: отношение произведения равновесных концентраций (или парциальных давлений) продуктов реакции, взятых в степенях их стехиометрических коэффициентов, к произведению для исходных веществ есть величина постоянная.Этот параметр называемая константой равновесия Кравн. Для обратимой гомогенной реакции: νAA + ν BB ó ν DD + ν F F константа равновесия может быть выражена в виде отношения равновесных молярных концентраций реагирующих веществ - ^ KC, равновесных парциальных давлений – KP, равновесных молярных долей -KX: Между константами равновесия KC, KP и KX для реакции с участием идеальных газов легко установить взаимосвязь, используя уравнение Менделеева – Клапейрона (pV=nRT) и закон Дальтона (pi = Xi p0 , где pi – парциальное давление i-ого газа, p0 – общее давление в системе, Xi – молярная доля i-ого газа): KP = KC(RT)Δν и KP = KX (p0)Δν (2), где Δν = (νD + νF ) – (νA + ν B) Константы KP , KC и KX называются эмпирическими константами равновесия, они могут быть численно равны, если Δν = 0, и иметь размерность, если Δν ≠ 0. Термодинамическая константа равновесия K0 безразмерна и может быть вычислена из стандартной энергии Гиббса: r G0T = - RT ln K0
| |||||||||||||||||