Пептид если от 10 до 40 аминокислот полипептид
 Скачать 7.45 Mb. Скачать 7.45 Mb.
|

99 вопросБиосинтез гема. Нарушение биосинтеза гема. Порфирии. Гем синтезируется во всех тканях, но с наибольшей скоростью в костном мозге и печени (рис. 13-2). В костном мозге гем необходим для синтеза гемоглобина в ретикулоцитах, в гепатоцитах - для образования цитохрома Р450. Первая реакция синтеза гема - образование 5-аминолевулиновой кислоты из глицина и сук-цинил-КоА (рис. 13-3) идёт в матриксе митохондрий, где в ЦТК образуется один из субстратов этой реакции - сукцинил-КоА. Эту реакцию катализирует пиридоксальзависимый фермент аминолевулинатсинтаза. 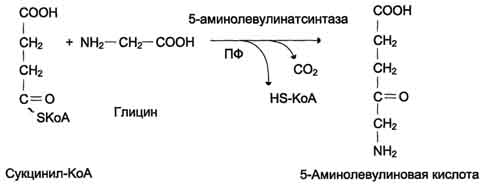 Из митохондрий 5-аминолевулиновая кислота поступает в цитоплазму. В цитоплазме проходят промежуточные этапы синтеза гема: соединение 2 молекул 5-аминолевулиновой кислоты молекулу порфобилиногена (рис. 13-4), 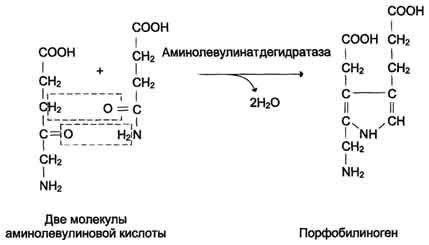 дезаминирование порфобилиногена с образованием гидроксиметилбилана, ферментативное превращение гидроксиметилбилана в молекулу уропор-фобилиногена III, декарбоксилирование последнего с образованием копропорфириногена III. Гидроксиметилбилан может также нефермента-тивно превращаться в уропорфириноген I, который декарбоксилируется в копропорфирино-ген I. Из цитоплазмы копропорфириноген III опять поступает в митохондрии, где проходят заключительные реакции синтеза гема. В результате двух последовательных окислительных реакций копропорфириноген III превращается в протопорфириноген IX, а протопорфириноген IX - в Протопорфирин IX. Фермент феррохела-таза, присоединяя к протопорфирину IX двухвалентное лентное железо, превращает его в гем (рис. 13-2). Источником железа для синтеза гема служит депонирующий железо белок ферритин. Синтезированный гем, соединяясь с α и β-полипепептидными цепями глобина, образует гемоглобин. Гем регулирует синтез глобина: при снижении скорости синтеза гема синтез глобина в ретикулоцитах тормозится. дезаминирование порфобилиногена с образованием гидроксиметилбилана, ферментативное превращение гидроксиметилбилана в молекулу уропор-фобилиногена III, декарбоксилирование последнего с образованием копропорфириногена III. Гидроксиметилбилан может также нефермента-тивно превращаться в уропорфириноген I, который декарбоксилируется в копропорфирино-ген I. Из цитоплазмы копропорфириноген III опять поступает в митохондрии, где проходят заключительные реакции синтеза гема. В результате двух последовательных окислительных реакций копропорфириноген III превращается в протопорфириноген IX, а протопорфириноген IX - в Протопорфирин IX. Фермент феррохела-таза, присоединяя к протопорфирину IX двухвалентное лентное железо, превращает его в гем (рис. 13-2). Источником железа для синтеза гема служит депонирующий железо белок ферритин. Синтезированный гем, соединяясь с α и β-полипепептидными цепями глобина, образует гемоглобин. Гем регулирует синтез глобина: при снижении скорости синтеза гема синтез глобина в ретикулоцитах тормозится.Регуляторную реакцию синтеза гема катализирует пиридоксальзависимый фермент аминолевулинатсинтаза. Скорость реакции регулируется аллостерически и на уровне трансляции фермента. Нарушения биосинтеза гема. Порфирии Наследственные и приобретённые нарушения синтеза гема, сопровождающиеся повышением содержания порфириногенов, а также продуктов их окисления в тканях и крови и появлением их в моче, называют порфириями ("порфирин" в переводе с греч. означает пурпурный). Наследственные порфирии обусловлены генетическими дефектами ферментов, участвующих в синтезе гема, за исключением аминолевулинатсинтазы. При этих заболеваниях отмечают снижение образования гема. Поскольку гем - аллостерический ингибитор аминолевулинатсинтазы, то активность этого фермента повышается, и это приводит к накоплению промежуточных продуктов синтеза гема - аминолевулиновой кислоты и порфириногенов. В зависимости от основной локализации патологического процесса различают печёночные и эритропоэтические наследственные порфирии. Эритропоэтические порфирии сопровождаются накоплением порфиринов в нормобластах и эритроцитах, а печёночные - в гепатоцитах. При тяжёлых формах порфирии наблюдают нейропсихические расстройства, нарушения функций РЭС, повреждения кожи. Порфириногены не окрашены и не флуоресцируют, но на свету они легко превращаются в порфирины. Последние проявляют интенсивную красную флуоресценцию в ультрафиолетовых лучах. В коже на солнце в результате взаимодействия с порфиринами кислород переходит в синглетное состояние. Синглетный кислород вызывает ускорение ПОЛ клеточных мембран и разрушение клеток, поэтому порфирии часто сопровождаются фотосенсибилизацией и изъязвлением открытых участков кожи. Нейропсихические расстройства при порфириях связаны с тем, что аминолевулинат и порфириногены являются нейротоксинами. Иногда при лёгких формах наследственных порфирии заболевание может протекать бессимптомно, но приём лекарств, являющихся индукторами синтеза аминолевулинатсинтазы, может вызвать обострение болезни. Индукторами синтеза аминолевулинатсинтазы являются такие известные лекарства, как сульфаниламиды, барбитураты, диклофенак, вольтарен, стероиды, гестагены. В некоторых случаях симптомы болезни не проявляются до периода полового созревания, когда повышение образования β-стероидов вызывает индукцию синтеза аминолевулинатсинтазы. Порфирии наблюдают и при отравлениях солями свинца, так как свинец нгибирует аминолевулинатдегидратазу и феррохелатазу. Некоторые галогенсодержащие гербициды и инсектициды являются индукторами синтеза аминолевулинатсинтазы, поэтому попадание их в организм сопровождается симптомами порфирии. 100 вопрос Гемоглобинопатии. Молекулярные основы серповидно-клеточной анемии. Талассемии. Гемоглобинопатии Серповидноклеточная анемия - тяжёлое наследственное заболевание, обусловленное точечной мутацией гена, кодирующего структуру β-цепи гемоглобина (см. раздел 4). В результате в эритроцитах больных присутствует HbS, β-цепи которого в шестом положении вместо гидрофильной глутаминовой кислоты содержат гидрофобную аминокислоту валин. Появление гидрофобной аминокислоты недалеко от начала молекулы способствует возникновению нового центра связывания, поэтому при низком парциальном давлении кислорода тетрамеры дезокси-HbS ассоциируют, образуя длинные микротрубчатые образования, которые полимеризуются внутри эритроцитов. Полимеризация приводит к нарушению структуры эритроцитов, они приобретают серповидную форму и легко разрушаются. При этом заболевании отмечают анемию, прогрессирующую слабость, отставание в развитии и желтуху. Носители гена серповидноклеточной анемии чаще всего встречаются среди африканского населения, так как они приобретают некоторое преимущество при заболевании малярией, часто встречающейся в странах с тропическим климатом. Причина сохранения гена серповидноклеточной анемии в популяции связана с тем, что в эритроцитах гетерозигот хуже развивается малярийный плазмодий, часть жизненного цикла которого проходит в эритроцитах человека. В связи с этим гетерозиготные носители дефектного .гена выживали при эпидемиях малярии, однако четверть их потомства погибала от серповиднок-леточной анемии. Талассемии - наследственные заболевания, обусловленные отсутствием или снижением скорости синтеза α- или β-цепей гемоглобина. В результате несбалансированного образования глобиновых цепей образуются тетрамеры гемоглобина, состоящие из одинаковых протомеров. Это приводит к нарушению основной функции гемоглобина - транспорту кислорода к тканям. Нарушение эритропоэза и ускоренный гемолиз эритроцитов и клеток-предшественников при талассемиях приводит к анемии. При β-талассемии не синтезируются β-цепи гемоглобина. Это вызывает образование нестабильных тетрамеров, содержащих только α-цепи. При этом заболевании в костном мозге из-за преципитации нестабильных α-цепей усиливается разрушение эритробластов, а ускорение разрушения эритроцитов в циркулирующей крови приводит к внутрисосудистому гемолизу. Как известно, для образования фетального гемоглобина р-цепи не требуются (см. раздел 4), поэтому клинически β-талассемия не проявляется до рождения, после чего происходит переключение синтеза HbF на НBА. В случае α-талассемии недостаток образования α-глобиновых цепей приводит к нарушению образования HbF у плода. Избыточные γ-цепи образуют тетрамеры, называемые гемоглобином Барта. Этот гемоглобин при физиологических условиях имеет повышенное сродство к кислороду и не проявляет кооперативных взаимодействий между протомерами. В результате гемоглобин Барта не обеспечивает развивающийся плод необходимым количеством кислорода, что приводит к тяжёлой гипоксии. При α-талассемии отмечают высокий процент внутриутробной гибели плода. Выжившие новорождённые при переключении с γ- на β-ген синтезируют β-тетрамеры или НBН, который, подобно гемоглобину Барта, имеет слишком высокое сродство к кислороду, менее стабилен, чем НBА и быстро разрушается. Это ведёт к развитию у больных тканевой гипоксии и к смерти вскоре после рождения. |