фазовые равновесия ФХ коллоквиум. Положительные и отрицательные отклонения от законов идеальных газов
 Скачать 1.83 Mb. Скачать 1.83 Mb.
|

1 2 Положительные и отрицательные отклонения от законов идеальных газов. Совершенные растворы характеризуются следующими свойствами: образование раствора не сопровождается тепловым эффектом (ΔH = 0), а изменение энтропии определяется уравнением (V.30). Реальные растворы, вообще говоря, не обладают этими свойствами и для них не соблюдается закон Рауля. На рис. V.8 и V.9 представлены зависимости давления пара от состава для двух реальных растворов. Прямые штриховые 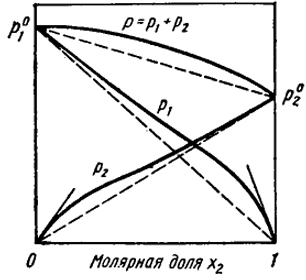 Рис. V.8. Положительные отклонения от закона Рауля. Касательные к кривым указывают области концентраций, где выполняются законы Генри и Рауля 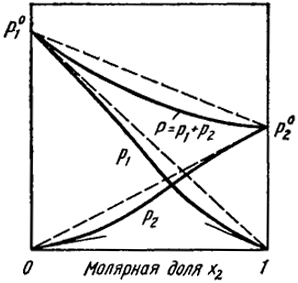 Ряс. V.9. Отрицательные отклонения от законов Рауля. Касательные к кривым указывают области концентраций, где выполняются законы Генри и Рауля линии соответствуют давлению пара, которое наблюдалось, если бы растворы были совершенными. На рис. V.8 показаны положительные отклонения, при которых давление пара выше, чем для совершенного раствора такой же концентрации, а на рис. V.9 - отрицательные отклонения, где давление пара меньше, чем для совершенного раствора. Наличие положительных отклонений показывает, что образование раствора затруднено по сравнению с совершенным. При больших положительных отклонениях жидкости расслаиваются. Отрицательные отклонения указывают на более сильную тенденцию к образованию раствора, чем в случае совершенного раствора. Это означает, что имеются дополнительные причины, облегчающие образование раствора. Так как изменение энергии Гиббса выражается уравнением ΔG = ΔH - TΔS, то отклонения в поведении реальных растворов от совершенных могут определяться как тем, что ΔH ≠ 0, так и тем, что изменение энтропии при смешении не подчиняется уравнению ΔSсов = -R[x ln x + (1 - x)ln(1 - х)]. Часто отклонения в поведении реальных растворов от идеальных законов обусловлены наличием теплоты смешения ΔH ≠ 0. В этом случае при положительных отклонениях имеет место поглощение тепла при образовании раствора, а при отрицательных - выделение тепла. Для описания термодинамических свойств реальных растворов используют различные приближенные теории. Согласно одной из них - теории регулярных растворов - отличие от свойств совершенных растворов обусловлено наличием теплоты смешения, т.е. тем, что ΔH ≠ 0. В то же время принимается, что изменение энтропии при образовании регулярного раствора остается таким же как у совершенного раствора идентичного состава и описывается уравнением (V.30). В гл. XII будет показано, что энтальпия образования бинарного регулярного раствора определяется выражением ΔH = Сx(1- х), где С- постоянная. Таким образом, с учетом уравнения (V.30) изменение энергии Гиббса при образовании регулярного раствора ΔG = ΔH - TΔS = Cx(1 - x) + RT[x ln x + (1 - x) ln (1 - x)]. (V.34) Если С > 0, т.е. раствор образуется с поглощением тепла, то величина ΔG более положительна, чем при образовании совершенного раствора той же концентрации. Это означает, что тенденция к самопроизвольному образованию раствора уменьшается по сравнению с совершенным и отклонения от закона Рауля положительны. В системах, где С > 0 и достаточно велика, первый член правой части уравнения (V.34) может по абсолютной величине превысить значение второго члена так, что ΔG > 0. В этом случае раствор не может образовываться. Возможно, однако, что он существует при больших Т, но при понижении температуры он будет расслаиваться. При С < 0 образование раствора сопровождается выделением тепла и отклонения от закона Рауля отрицательны. Растворы металлов, особенно переходных, не описываются теорией регулярных растворов. В частности, в бинарных сплавах не наблюдается концентрационная симметричность термодинамических свойств (ΔG, ΔH и ΔS), которая вытекает из уравнения (V.34). Методы определения активности компонентов раствора. Экспериментальные способы измерения активности основаны на изучении реакций между изучаемым компонентом в растворе и какой-либо второй фазой, в которой активность этого компонента может быть легко определена, например, с идеальным газом или чистым веществом. При равновесии химические потенциалы и активности компонента в обеих фазах одинаковы. Часто задача сводится к нахождению активности во второй фазе. Для этого используют следующие методы. Измерение давления пара. Этот метод, состоящий в измерении парциального давления пара компонента над раствором pi и над чистым веществом p0i, непосредственно приводит к нахождению аi в соответствии с определением этой величины [уравнение (21)]. Применение радиоактивных изотопов позволило проводить такие измерения в металлических системах, где давления паров весьма малы даже при относительно высоких температурах. Используемые методы основаны на том, что величины pi пропорциональны скорости сублимации или испарения vi вещества в вакууме. Эту величину находят либо по убыли массы образца после выдержки в вакууме при постоянной и достаточно высокой температуре, либо по определению количества испарившегося вещества, сконденсировавшегося на мишени. Активность рассчитывают по уравнению аi=vi/v0i где v0i — скорость сублимации чистого вещества. Измерение электродвижущих сил (э. д. с.). Метод основан на определении работы обратимого, изотермического переноса компонента i из чистого состояния в сплав. Такой процесс осуществляют в гальваническом элементе, в котором один электрод — чистый металл г, а другой — сплав i с более благородным элементом, например металлом. Электролитом служит ионный проводник, содержащий катионы г, которые могут в нем мигрировать. Так как свободная энергия (химический потенциал μ0i) чистого вещества больше соответствующей величины μi в растворе, то при замыкании элемента возникает самопроизвольный перенос i от одного электрода к другому через электролит. Благодаря освобождающейся при этом энергии элемент производит работу А, равную произведению электродвижущей силы E на заряд катионов п и на число Фарадея F. (96493 Кулона), т. е. A = nEF. Эта работа равна убыли свободной энергии при рассматриваемом процессе А = -ΔG = nEF или разности химических потенциалов чистого г и г в растворе согласно уравнению Таким образом: т. е. измерение э. д. с. позволяет непосредственно определить активность. Рассмотрим схему гальванического элемента для определения активности алюминия, растворенного в твердом никеле: На левом электроде идет реакция растворения алюминия: Al (т)→Al5++3е, при которой этот электрод заряжается отрицательно, так как на него переходят три электрона (е). На правом электроде совершается реакция восстановления: Al3++3е→Al (раствор в никеле). Сумма этих двух электродных реакций описывает процесс переноса, происходящий в элементе: Al (т)→Al (раствор в никеле). Так как заряд иона алюминия равен 3, то n=3 и активность алюминия в никеле вычисляют по уравнению RT In аAl = -3 EF. Изучение распределения вещества между двумя фазами. Сплав, содержащий компонент i при заданной температуре, приводят в равновесие с раствором, для которого известна зависимость между активностью аi и концентрацией. Таким раствором, который служит фазой сравнения, может быть, например, разбавленный раствор, где аi∞=γ0iCi. При равновесии в растворе устанавливается концентрация Ci, а в сплаве Ci. Так как активности i в этом состоянии в обеих фазах одинаковы, то где f'i — коэффициент активности i в сплаве. Таким образом, величина f'i=γ0i(Ci/Ci') находится из измерений отношений Ci/Ci', т. е. из данных о распределении вещества между двумя фазами. Определяя fi' в зависимости от Ci', можно найти область концентраций, в которой эта величина становится постоянной, т. е. найти границу области закона Генри для изучаемого сплава. В этой области отношение Ci/Ci', называемое коэффициентом распределения, не зависит от концентрации. К описанному методу (изучение распределения между фазами) весьма близок и способ нахождения активности из диаграмм состояний или просто из данных о растворимости. Он основан на уже отмечавшемся равенстве активностей компонента в двух соприкасающихся фазах при равновесии. Это позволяет из данных об активности компонента в одной фазе и по диаграмме состояния найти активность этого же компонента в другой фазе. В простейшем случае, если из жидкого или твердого раствора выделяется чистое вещество, активность которого равна единице, очевидно, активность компонента в растворе (насыщенном) тоже равна единице. Это позволяет найти коэффициент активности из уравнения Изучение химических равновесий. Активность компонента i в данной фазе определяется в результате изучения какой-либо реакции этого компонента с веществами, находящимися в другой фазе. Так, активность углерода, растворенного в железе, может быть найдена из данных о реакции С + 2Н2 (г) = CH4 (г) (VI), константа равновесия которой где для краткости pCH4/p2H2=r. Сначала примем в качестве стандартного состояния графит. При этом Ka находят путем измерения отношения PCH4/p2H2 при равновесии с графитом, которое в этом случае обозначают через r0. Так как в стандартном состоянии аC=1, то из уравнения (44) следует, что Ka=r0. Отсюда для ненасыщенного раствора активность углерода вычисляют по уравнению а коэффициент активности — по уравнению γC=aC/NC, где NC — атомная доля углерода в железе. Для рассматриваемой системы в литературе часто выражают концентрацию в процентах по массе, а в качестве состояния сравнения используют бесконечно разбавленный раствор. В этом случае экспериментально определяемую при равновесии величину выражают уравнением Опыт показывает, что для твердых растворов на основе γ-Fe величина Kэ при концентрациях углерода выше 0,2—0,3% (по массе) не является постоянной и зависит от концентрации, т. е. в этой области не выполняется закон Генри. Поэтому для того, чтобы найти истинную константу равновесия для выбранного стандартного состояния Ka∞: определяют K3 при различных концентрациях углерода (при постоянной температуре). Полученные значения экстраполируют в сторону малых концентраций углерода вплоть до нулевой, вблизи которой аС∞=[С] и fС=1 и, следовательно, Ka∞=Kэ (при [С]→0). Обычно такую экстраполяцию проводят графически, откладывая lgKэ в зависимости от [С]. В результате сопоставления уравнений (45) и (47) и исключения из них r получаем соотношение между величинами активностей, определенных относительно двух разных, стандартных состояний: где NC — атомная доля. По линиям двухфазного равновесия можно найти активность в одной фазе, если для данного компонента она известна в другой фазе, при помощи соотношения, аналогичного уравнению (45). Например, по известной активности углерода в феррите (где соблюдается закон Генри) можно определить активность углерода в равновесном аустените. Определение активности одного компонента по известной активности другого компонента. Способ основан на том, что активности компонентов, находящихся в одном растворе взаимно, зависимы друг от друга. Изменение активности одного из них вследствие уменьшения или увеличения его концентрации влечет за собой изменения активности и других компонентов. Активности компонентов связаны между собой определенным уравнением (уравнение Гиббса—Дюгема). В случае бинарного раствора оно имеет вид где индексы 1 и 2 относятся к двум компонентам раствора. Для расчетов, однако, удобнее аналогичное уравнение для коэффициентов активности: При интегрировании этого уравнения в пределах между двумя концентрациями N2 и N'2 (N2≥N'2) получим Зная зависимость γ2 от состава, т. е. от N2/N1, можно найти величину интеграла в правой части уравнения (обычно это осуществляют графически). Для определения γ1 при концентрации N2 необходимы данные о значениях γ2 при достаточно низких концентрациях вблизи N'2, т. е. в области, где N'2→0, γ1'→1 (ln γ1'→0). Величину активности находят из известного соотношения a1=γ1N1. Условия термодинамического равновесия между фазами. В наиболее общей форме условия фазового равновесия можно получить, исходя из первого и второго начал термодинамики. Второе начало термодинамики в формулировке Клаузиуса дает ключ для получения условий равновесия: при равновесии энтропия изолированной системы максимальна. Это означает, что все самопроизвольные процессы, протекающие в изолированной системе, сопровождаются увеличением энтропии. Например, диффузия атомов и химические реакции ведут к увеличению энтропии. Рассмотрим вопрос о том, какое из возможных состояний системы отвечает термодинамическому равновесию, т. е. при отсутствии внешнего воздействия может существовать неограниченно долго. Для равновесия недостаточно, чтобы какой-либо термодинамический потенциал имел экстремальное значение. Принцип экстремальности применим только в том случае, когда сравниваемые состояния удовлетворяют определенным условиям: например, рассматриваются состояния изолированной системы или состояния с постоянной температурой и давлением. В изолированной системе при равновесии экстремального значения достигает энтропия. Однако гораздо чаще процессы протекают не в изолированной системе, а при постоянной температуре. Рассмотрим условие изотермического равновесия. Для таких систем справедливо основное неравенство термодинамики для неравновесных процессов
Если рассматривать изотермические процессы, протекающие при постоянном объеме, и перейти к независимым переменным, объему и температуре (V и T), то уравнение (13.8) примет вид
Следовательно, в рассматриваемой системе с неизменным количеством вещества могут протекать лишь такие процессы, при которых свободная энергия Гельмгольца не растет. Они прекращаются, как только свободная энергия F достигает минимума, что соответствует термодинамическому равновесию системы. Таким образом, при постоянных температуре (T) и объеме (V) состоянию равновесия отвечает минимум свободной энергии Гельмгольца
В этом случае равновесному состоянию системы отвечает минимум термодинамического потенциала Гиббса Найденные условия термодинамического равновесия позволяют выделить равновесное состояние среди других, но они ничего не говорят о возможных внутренних условиях, определяющих равновесие между компонентами системы. Исследуем условия равновесия фаз, имеющих в общем случае различный химический состав. Зафиксируем опять температуру (T) и давление (p). Тогда для системы из двух фаз α и β должно выполняться требование минимума термодинамического потенциала. В состоянии равновесия имеем
Поскольку температура, давление и общее количество вещества неизменны, то изменения возможны только в отношении состава фаз, т. е. какое-то количество компонента K может перейти из одной фазы в другую. При этом число молей компонента K в фазах α и β:
Это условие должно выполняться для любого изменения количества вещества
Итак, при равновесии двух фаз химические потенциалы имеют одинаковую величину для каждого из компонентов, входящих в состав обеих фаз. Связь между температурой и давлением при фазовом переходе. (Уравнение Клапейрона-Клаузиуса) Уравнение Клапейрона — Клаузиуса — термодинамическое уравнение, относящееся к квазистатическим (равновесным) процессам перехода вещества из однойфазы в другую (испарение, плавление, сублимация, полиморфное превращение и др.). Согласно уравнению, теплота фазового перехода (например, теплота испарения, теплота плавления) при квазистатическом процессе определяется выражением где Уравнение названо в честь его авторов, Рудольфа Клаузиуса и Бенуа Клапейрона. Элементарный вывод 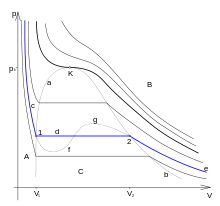 Между температурой фазового перехода и внешним давлением существует функциональная связь, причём при фазовом переходе производная 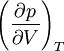 терпит разрыв. Тогда изотермы для рассматриваемого вещества будут иметь характерный вид, изображённый на рисунке. Для вывода существенен горизонтальный участок изотермы, соответствующий фазовому переходу. Слева и справа от этого участка всё вещество находится в одной фазе. Осуществим цикл Карно при бесконечно малой разности температур следующим образом: сначала сообщаем телу теплоту, переводя его из состояния 1 в состояние 2, затемадиабатически охлаждаем его на температуру dT, после чего замыкаем цикл, отводя теплоту и переводя вещество в фазу 1 с последующим адиабатическим нагревом. Совершённая работа равна площади цикла: терпит разрыв. Тогда изотермы для рассматриваемого вещества будут иметь характерный вид, изображённый на рисунке. Для вывода существенен горизонтальный участок изотермы, соответствующий фазовому переходу. Слева и справа от этого участка всё вещество находится в одной фазе. Осуществим цикл Карно при бесконечно малой разности температур следующим образом: сначала сообщаем телу теплоту, переводя его из состояния 1 в состояние 2, затемадиабатически охлаждаем его на температуру dT, после чего замыкаем цикл, отводя теплоту и переводя вещество в фазу 1 с последующим адиабатическим нагревом. Совершённая работа равна площади цикла:Сообщённая теплота равна где Отсюда 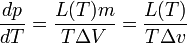 Зависимость давления насыщенного пара жидкости и твердого тела от температуры. Состояние насыщенного пара, как показывает опыт (мы говорили об этом в предыдущем параграфе), приближенно описывается уравнением состояния идеального газа (10.4), а его давление определяется формулой С ростом температуры давление растет. Так как давление насыщенного пара не зависит от объема, то, следовательно, оно зависит только от температуры. Однако зависимость рн.п. от Т, найденная экспериментально, не является прямо пропорциональной, как у идеального газа при постоянном объеме. С увеличением температуры давление реального насыщенного пара растет быстрее, чем давление идеального газа (рис.11.1, участок кривой АВ). Это становится очевидным, если провести изохоры идеального газа через точки А и В (штриховые прямые). Почему это происходит?  При нагревании жидкости в закрытом сосуде часть жидкости превращается в пар. В результате согласно формуле (11.1) давление насыщенного пара растет не только вследствие повышения температуры жидкости, но и вследствие увеличения концентрации молекул (плотности) пара. В основном увеличение давления при повышении температуры определяется именно увеличением концентрации. Главное различие в поведении идеального газа и насыщенного пара состоит в том, что при изменении температуры пара в закрытом сосуде (или при изменении объема при постоянной температуре) меняется масса пара. Жидкость частично превращается в пар, или, напротив, пар частично конденсируется. С идеальным газом ничего подобного не происходит. Когда вся жидкость испарится, пар при дальнейшем нагревании перестанет быть насыщенным и его давление при постоянном объеме будет возрастать прямо пропорционально абсолютной температуре (см. рис.11.1, участок кривой ВС). Фазовые переходы второго рода. При фазовых переходах второго рода химические потенциалы
Следовательно, в точке фазового перехода II рода непрерывны не только термодинамический потенциал, но и его первые производные по температуре или давлению (энтропия и объем), тогда как вторые производные терпят разрыв (изменяются скачком). Скачкообразно изменяются и величины, выражающиеся через вторые производные: теплоемкость при постоянном давлении
Эти величины могут претерпевать скачок и при фазовых переходах первого рода. Поскольку при фазовых переходах II рода энтропия меняется непрерывно, то отсутствует теплота перехода (13.20). Особенность фазовых переходов II рода состоит в невозможности перегрева или переохлаждения фаз: каждая фаза существует только в своем температурном интервале. Высокотемпературное состояние в области Фазовые переходы II рода могут проходить по разным механизмам за счет: • незначительного смещения атомов в решетке; • изменения степени упорядоченности атомов в кристаллической фазе (переходы типа «порядок – беспорядок»); • перехода вещества из ферромагнитного состояния в парамагнитное; • перехода металлов из обычного в сверхпроводящее состояние. 1)Гетерогенные равновесия:Если внутри системы нет поверхностей раздела, отделяющих различные по составу части системы, то эта система называется гомогенной. гетерогенной называется система состоящая из нескольких фаз, различающихся по строению или химическому составу.Условием равновесия в гетерерогенной системе является равенство химических потенциалов каждого компонента во всех фазах системы ( 2)Основные понятия и определения:Фазой называется часть гетерогенной системы, ограниченная поверхностью раздела и характеризующаяся одинаковыми во всех точках или непрерывно меняющимися от точки к точке физическими и химическими свойствами.Жидкие и тверды Каждая система состоит из одного или нескольких веществ. Индивидуальные химические вещества, которые могут быть выделены из системы и существовать вне ее самостоятельно в виде отдельной фазе, называется составляющими веществами системы(составляющая часть системы). Если между составляющими веществами нет химического взаимодействия, то состав каждой фазы системы при любых условиях однозначно выражается через концентрации составляющих веществ, и в этом случае они называются компонентами.По числу компонентов системы бывают(одно-,двукомпонентные,тройные)Наименьшее число,составляющих систему достаточное для образования всех ее фаз называетсяся число независ.компонентов системы. В случае химического взаимодействия между составляющими веществами число компонентов может быть меньше чем число составляющих веществ. При одном и том же числе составляющих веществ число компонентов может быть разным. В этом случае число компонентов равняется наименьшему числу составляющих веществ системы, достаточных для определения состава любой фазы систем. В случае равновесия между составляющими веществами существует дополнительное уравнение взаимосвязи между их концентрациями, которое уменьшает число веществ, концентрации которых необходимо знать для описания системы.Таким образом,число компонентов равняется числу составляющих веществ системы минус число уравнений, связывающих равновесные концентрации этих веществ. термодинамические степени свободы-Независимые термодинамические параметры, определяющие состояние каждой фазы равновеснойсистемы. .По числу степеней свободы системы бывают (безвариантные,одно-,би-,трехвариантные,поливариантные) 3)Правило фаз Гиббса:Уравнение, выражающее зависимость числа степеней свободы от числа компонентов и числа фаз, находящихся в равновесии, называется правилом фаз Гиббса С = К – Ф + n Таким образом, можно сказать, что число степеней свободы равновесной термодинамической системы равно числу компонентов минус число фаз плюс число внешних факторов оказывающих влияние на систему. 5)Применение правила фаз для анализа однокомпонентных систем:Для всех однокомпонентных систем внешними факторами которых являются T и P,влияющими на компоненты системы С=3-Ф Отсюда следует, что максимальное число фаз в однокомпонентной системе, достигаемое при минимальной (нулевой) её вариантности, равно трём; ни давление, ни температуру для трёхфазной однокомпонентной системы задать произвольно нельзя. На фазовой диаграмме сосуществованию трёх фаз соответствует тройная точка с фиксированными значениями давления и температуры. При всякой другой температуре или другом давлении равновесие трёх фаз невозможно: в системе будут происходить изменения, в результате которых одна или две фазы исчезнут. 8)Энантиотропные и монотропные превращения: Переход одной аллотропной модификации в другую происходит при изменении температуры или давления (или одновременном воздействии обоих факторов) и связан со скачкообразным изменением свойств вещества. Этот процесс бывает обратимым (энантиотропным) и необратимым (монотропным). Примером энантиотропного перехода может служить превращение ромбической серы в моноклинную α-S (ромб.) ↔ β-S (монокл.) при 95,6 °C. При обычной температуре стабильной является ромбическая модификация серы, которая при нагревании до 95,6 °С при нормальном давлении переходит в моноклинную форму. Последняя при охлаждении ниже 95,6 °С вновь переходит в ромбическую форму. Таким образом, переход одной формы серы в другую происходит при одной и той же температуре, и сами формы называются энантиотропными. К монотропному переходу относится превращение белого фосфора P4 под давлением 1,25 ГПа и температуре 200 °C в более стабильную модификацию — чёрный фосфор. При возвращении к обычным условиям обратный переход не происходит. Переход из нестабильной формы в стабильную в принципе возможен при любой температуре, а обратный — нет, то есть определенная точка перехода отсутствует. Три известные модификации олова переходят друг в друга различным образом. При обычных условиях устойчиво β-Sn (пластичное белое олово) с тетрагональной кристаллической решеткой. Выше 173 °С β-Sn энантиотропно превращается в хрупкую модификацию γ-Sn, а ниже 13,2 °C β-Sn переходит монотропно в порошкообразное α-Sn (серое олово) с кубической решёткой типа алмаза. Этот полиморфный переход происходит с малой скоростью, но резко ускоряется в контакте с серым оловом — плотные куски белого олова рассыпаются в пыль («оловянная чума»). Обратный процесс возможен только путем переплавки. 7)Диаграмма состояния серы: 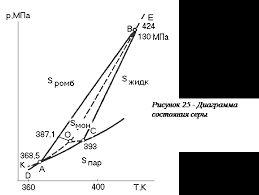 Примером энантиотропного перехода может служить превращение ромбической серы в моноклинную α-S (ромб.) ↔ β-S (монокл.) при 95,6 °C. При обычной температуре стабильной является ромбическая модификация серы, которая при нагревании до 95,6 °С при нормальном давлении переходит в моноклинную форму. Последняя при охлаждении ниже 95,6 °С вновь переходит в ромбическую форму. Таким образом, переход одной формы серы в другую происходит при одной и той же температуре, и сами формы называются энантиотропными Диаграмма состояния воды: Диаграмма состояния воды — система с одним компонентом H2O, поэтому наибольшее число фаз, которые одновременно могут находиться в равновесии, равно трем. Эти три фазы — жидкость, лед, пар. Число степеней свободы в этом случае равно нулю, т.е. нельзя изменить ни давление, ни температуру, чтобы не исчезла ни одна из фаз. Обычный лед, жидкая вода и водяной пар могут существовать в равновесии одновременно только при давлении 0,61 кПа и температуре 0,0075°С. Точка сосуществования трех фаз называется тройной точкой (O). 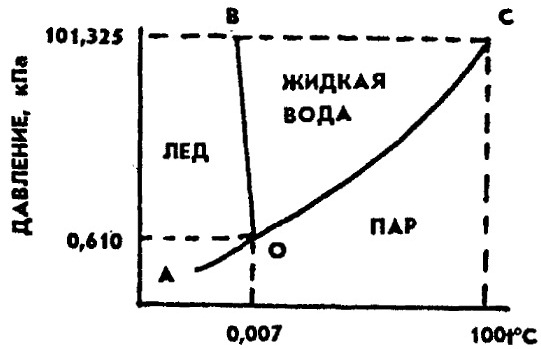 Кривая ОС разделяет области пара и жидкости и представляет собой зависимость давления насыщенного водяного пара от температуры. Кривая ОС показывает те взаимосвязанные значения температуры и давления, при которых жидкая вода и водяной пар находятся в равновесии друг с другом, поэтому она называется кривой равновесия жидкость—пар или кривой кипения. Кривая ОВ отделяет область жидкости от области льда. Она является кривой равновесия твердое состояние—жидкость и называется кривой плавления. Эта кривая показывает те взаимосвязанные пары значений температуры и давления, при которых лед и жидкая вода находятся в равновесии. Кривая OA называется кривой сублимации и показывает взаимосвязанные пары значений давления и температуры, при которых в равновесии находятся лед и водяной пар. 4)Общие представления о диаграммах состояния: Диаграмма состояния (фазовая диаграмма), графическое изображение всех возможных состояний термодинамической системы в пространстве основных параметров состояния - температуры Т, давления р и состава х (обычно выражаемого молярными или массовыми долями компонентов). Для сложных систем, состоящих из многих фаз и компонентов, построение диаграмма состояния является единственным методом, позволяющим на практике установить, сколько фаз и какие конкретно фазы образуют систему при данных значениях параметров состояния. Каждое реально существующее состояние системы на диаграмме состояния изображается так называемой фигуративной точкой; областям существования одной фазы отвечают участки пространства (на трехмерных диаграммах состояния) или плоскости (на двухмерных диаграммах состояния), условиям сосуществования фаз соответствуют поверхности или линии; изменение фазового состояния системы рассматривается как движение фигуративной точки на диаграмме состояния. Анализ относительного расположения объемных участков, поверхностей, линий и точек, которые образуют диаграмму состояния, позволяет однозначно и наглядно определять условия фазового равновесия, появления в системе новых фаз и химических соединений, образования и распада жидких и твердых растворов и т. п. Теоретическими основами построения и интерпретации диаграмм состояния равновесных систем являются: 1) условие фазового равновесия, согласно которому химические потенциалы μi каждого i-го компонента во всех фазах при равновесии равны; 2) условие химического равновесия, согласно которому сумма химических потенциалов вступающих в реакцию веществ при равновесии равна аналогичной сумме для продуктов реакции; 3) фаз правило Гиббса, согласно которому число компонентов К, число фаз Ф ивариантность системы С (т.е. число независимых параметров состояния, которые можно в определенных пределах изменять без изменения числа и природы фаз) связаны соотношением: С = К — Ф + 2. Цифра 2 означает, что учитываются только два интенсивных параметра состояния - температура и давление. Если учитываются и другие параметры, например, напряженности электромагнитного или гравитационного полей, вариантность системы соответственно увеличивается.Безвариантные(с=0),одно-(с=1),би-(с=2),трехвариантные(с=3),поливариантны(с> 4). 4)правило о соприкасающихся пространствах состояния, согласно которому если два разных пространства состояния (поля в случае плоской диаграммы) соприкасаются по линии, то они различаются между собой на одну фазу, если поля соприкасаются в точке, то состояния различаются на две фазы.  Состав раствора и состав пара Состав пара, как правило, отличается от состава раствора. Соотношение между составом жидкости и пара определяется первым законом Коновалова: «Пар по сравнению с жидкостью, находящейся с ним в равновесии, обогащен тем компонентом, добавление которого к жидкости повышает общее давление над ней (или понижает температуру кипения)». Другими словами: «Пар обогащен по сравнению с жидкостью более летучим компонентом». Из двух жидкостей более летучей является та, которая имеет более низкую температуру кипения или, другими словами, та, над которой выше давление насыщенного пара. Так, если взять жидкую смесь воды и ацетона с равными концентрациями компонентов, то в парах будет содержаться больше ацетона, т.к. давление насыщенного пара над ацетоном больше, чем над водой. Второй закон Коновалова описывает растворы с отклонениями от свойств идеальных растворов и объясняет существование азеотропных растворов, состав которых при перегонке не изменяется: «Экстремумы на кривых полного давления пара отвечают такому равновесию раствора и насыщенного пара, при котором состав обеих фаз одинаков». Азеотропные смеси (азеотропы) - жидкие смеси, характеризующиеся равенством составов равновесных жидкой и паровой фаз. При их перегонке образуется конденсат того же состава, что и исходный раствор; поэтому азеотропные смеси называются также нераздельнокипящими. Наличие азеотропных смесей существенно затрудняет разделение жидких смесей и требует применения специальных методов ректификации. 1 2 |
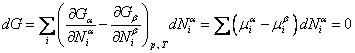 .
.