Рвотные и противорвотные препараты. рвотные и противорвотные лекарственные препараты
 Скачать 2.27 Mb. Скачать 2.27 Mb.
|

5.2 Синтез противорвотных лекарственных средств5.2.1 Синтез аминазина 2-Хлорфенотиазин – основной полупродукт синтеза ценного и широко применяемого лечебного препарата аминазина [19]. Кроме того, он является основным полупродуктом синтеза другого лечебного препарата – хлорацизина, применяемого при лечении заболеваний сердечно-сосудистой системы. Наиболее рациональноной схемой получения 2-хлорфенотиазина является конденсация 3-хлордифениламина с серой (сплавление) в присутствии йода.  Реакция протекает с образованием двух изомерных хлорфенотиазинов – 2-хлорфенотиазина и 4-хлорфенотиазина. Впервые эта реакция описана Шарпантье и Жолио. Авторы проводили конденсацию при небольшом избытке серы в присутствии 1 % йода при 180°С. Они приводят данные о температуре плавления образующихся продуктов без описания метода разделения и указания выхода 2-хлорфенотиазина. Н. В. Савицкая и соавторы разработали условия конденсации 3-хлордифениламина с серой и очистки плава хлорфенотиазина. Реакцию 3-хлордифениламина с небольшим (2 % теоретического количества) избытком серы проводят в присутствии йода (1 % веса 3-хлордифениламина). Реакционную массу нагревают до 175–180°С и выдерживают при этой температуре до прекращения выделения сероводорода. Плав после кипячения в хлорбензоле с активированным углем фильтруют. Выкристаллизовавшийся и отфильтрованный хлорфенотиазин промывают хлорбензолом и спиртом. Выход 2-хлорфенотиазина составляет 56,2 %, считая на 3-хлордифениламин. Температура плавления 2-хлорфенотиазина 191–194°С. В производстве аминазина получение 2-хлорфенотиазина является наиболее трудным процессом, что обуловлено относительно высокой температурой сплавления (175–180°С), образованием изомера – 4-хлорфенотиазина, смолистых веществ и выделением больших количеств сероводорода. В производственных условиях в силу ряда причин осмоление более значительно, чем в лабораторных условиях, поэтому даже сравнительно невысокий выход 2-хлорфенотиазина (56 %) в производстве практически не достигается. Все эти обстоятельства заставили нас искать путей смещения равновесия реакции с целью повышения выхода 2-хлорфенотиазина. Попытки вести реакцию в каких-либо наиболее доступных и обычно применяемых органических растворителях не привели к положительным результатам. Лучшей средой для ведения реакции оказалось само основное вещество – 3-хлордифениламин. Мы установили, что проведение реакции 3-хлордифениламина с серой при избытке 3-хлордифениламина и в присутствии остатка после отделения 2-хлорфенотиазина, содержащего главным образом 4-хлорфенотоазин, смещает реакцию в сторону преомущественного образования 2-хлорфенотиазина. Экспериментальная часть В трехгорловую круглодонную колбу, снабженную мешалкой и термометром, помещают 100 г перегнанного 3-хлордифениламина (0,49 г/м), 20 г серы (0,31 г/м) и 1 г йода (0,005 г/м). Полученную смесь нагревают при перемешивании до 170–180°С и выдерживают при этой температуре до прекращения выделения сероводорода (1 – 1½ часа). По окончании выдержки к реакциовной массе прибавляют 175 мл хлорбензола и 5 г угля, кипятят в течение 15 минут (130–132°С) и затем фильтруют горячий раствор, освобождая его от угля и смолистых примесей. Фильтрат охлаждают до 5–10°С и оставляют на 3–4 часа для кристаллизации. Вьипавший осадок 2-хлорфенотиазина отфильтровывают, промывают небольшим количеством хлорбензола и этилового спирта и сушат при 100°С. Получают 42–45 г 2-хлорфенотиазина с температурой плавления 195–200°С (в пределах 1°С). От маточного раствора после отделения 2-хлорфенотиазина отгоняют спирт и хлорбензол, к остатку прибавляют 32 г 3-хлордифениламина, 10 г серы и 0,5 г йода и повторяют синтез, как описано выше. Подобным же образом поступают с остатками после отделения 2-хлорфенотиазина в последующих 9–10 опытах с той только разницей, что нагревают до 175–180°, а не до 150–160°, как в первых двух опытах. В итоге при употреблении в цикле из 11 опытов 420 г 3-хлордифенил-амина получают 380 г 2-хлорфенотиазина, что составляет 79 % теоретического выхода, считая на израсходованный 3-хлордофениламин. Количество 3-хлордифениламина (32 г) во втором и последующих опытах определяется весом получаемых остатков после отделения 2-хлорфенотиазина с учетом того, чтобы сумма веса 3-хлордифениламина и остатка была близка к первоначальной загрузке, т. е. к 100 г. Поскольку в цикле находится избыток 3-хлордифениламина, количество взятой серы близко к теоретическому (по отношению к вновь загружаемому 3-хлордифениламину). Вес остатка не увеличивается с увеличением числа проведенных опытов, что подтверждает предположение о смещении равновесия реакции в сторону образования 2-хлорфенотиазина, когда в остатке уже имеется изомер 4-хлорфенотиазин, так как вновь загружаемый 3-хлордифениламин расходуется главным образом на образование 2-хлорфенотиазина, иначе с увеличением числа проведенных опытов должен был бы существенно увеличиваться вес остатка. Рециркуляция остатка длится до тех пор, пока не истощится избыток 3-хлордифениламина. При 10-й рециркуляции остатка 2-хлорфенотиазин имеет точку плавления 193–194°С, окраска хлорфенотиазина темно-зеленаяж. Дальнейшая рециркуляция уже нецелесообразна, так как наступает еще более резкое понижение качества. Мы провели серию опытов, в которой, начиная с 3-го опыта, брали по 35 г 3-хлордифениламина т. е. на 10 % больше, чем в предыдущих опытах. В этом случае удалось вести рециркуляцию остатка до 20-го опыта, но выход остался на прежнем уровне (77 %). Поэтому соотношение компонентов реакции в первой серии опытов следует считать наиболее рациональным. Описанный способ позволяет получать 2-хлорфенотиазии высокого качества, с температурой плавления 195–200°С (в пределах 1°С) и выходом 79 % теоретического, считая на 3-хлордифениламин. Заключительной стадией в синтезе аминазина (I*HCl) является алкилирование 2-хлорфенотиазина (II) диметиламинохлорпропаном (III) [20]. Подобран оптимальный режим процесса: реагенты II и III кипятят в смеси толуола с хлорбензолом в присутствии порошкообразного едкого натра, причем выход I достигает 90 %. Проведение реакции аналогичным образом, но в присутствии катализатора межфазного переноса (КМФ) не дает очевидных преимуществ; выход I составляет 78 %. В более мягких условиях, типичных для реакции межфазного переноса (80°С, 25 мол.% КМФ, бензол/водный раствор NaOH) соединение I получить не удалось, хотя в тех же условиях фенотиазин II алкилируется хлористым бензилом и бромистым этилом. Низкая эффективность III как алкилирующего агента была отмечена, но не получила объяснения.  I: R = (CH2)3NMe; II: R = H; IV: R = CH2Ph; V: R = Me; VII: R = CH2CH=CH2 Экспериментальная часть ГЖХ-анализы проведены на хроматографах «Varian-3700» (США) и «Chrom-5» (ЧССР) с пламенно-ионизационным детектором; колонки стеклянные или стальные (100×0,3 см), заполненные сорбентом (5% OV-17) на хроматоне «N-super». Замена стеклянных колонок на металлические, а также изменение температуры испарителя (210–350°С) не приводит к термокаталитическому разложению основания аминазина I: на хроматограммах не меняется соотношение пиков и не появляются дополнительные пики. Режим анализа: температура анализа колонок 210°С, испарителя 250°С, детектора 250°С, скорость газа-носителя (азот) 30 мл/мин, время удерживания веществ V, VII и I 3, 3,8 и 7 соответственно. Масс-спектры электронного удара получены на хромато-масс-спектрометре МАТ-112 («Varian», ФРГ), энергия ионизирующих электронов 70 эВ. Ввод образца реакционной массы осуществлялся через хроматограф «Varian Aerograph 1440»; колонки и режим их работы такие же, как и при ГЖХ-анализе. Гидрохлорид 2-хлор-10-(γ-диметиламинопропил)-фенотиазина (I*HCl) и 2-хлор-10-метилфенотиазин (V). Реакцию II (0,05 моль) и III проводят согласно [1], продолжительность отгонки воды 4,5 ч. Далее реакционную смесь промывают водой, смесь растворителей отгоняют в вакууме, остаток сушат до постоянной массы (16,13 г) и анализируют методом ГЖХ. Полученную смесь растворяют в 70 мл толуола, прибавляют раствор HCl в этаноле, раствор упаривают в вакууме до объема 30 мл, выпавшие кристаллы отфильтровывают и после перекристаллизации из смеси толуол–изопропиловый спирт (4:1) получают 10,92 г I*HCl (61,4%). Маточный раствор после отделения технического I*HCl промывают водой, толуол отгоняют, остаток тщательно сушат в вакууме и анализируют на хромато-масс-спектрометре. Масс-спектр m/z (IОТН): V – 247(100), 232(65), 215(12), 212(19); VII – 273(11), 247(8), 233(95), 232(100), 198(39). После двух перекристаллизаций из гексана получают 0,1 г V, т.пл. 81–83°С. 5.2.2 Синтез анестезина 5.2.2.1 Синтез анестезина из р-нитротолуола В качестве исходного сырья для получения этого продукта лучше всего применять р-нитротолуол, во-первых, потому, что его легко и дешево можно купить, и, во-вторых, потому, что отдельные фазы получения через этиловый эфир р-нитробензойной кислоты до эфира р-аминобензойной кислоты дают хорошие выхода и протекают гладко, хотя и требуют частично довольно продолжительного времени для завершения реакции [21]:  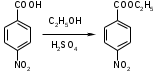  р-Нитробензойная кислота из р-нитротолуола. В гомогенно освинцованном двустенном котле с крышкой, мешалкой, обратным холодильником и трубкой для выдавливания большого внутреннего диаметра заливают 200 л воды, к которым при помешивании и охлаждении водой медленно приливают 150 кг технической серной кислоты 66° Вё. По охлаждении прибавляют 60 кг двухромовокислого калия и 15 кг р-нитротолуола, закрывают люк и нагревают до кипения 24 часа при помешивании с обратным холодильником. Перемешивают для того, чтобы нитробензойная кислота не обволакивала нитротолуола и не мешала окислению. Кипение способствует окислению; при температуре ниже кипения окисление протекает медленнее. Охлаждающая вода в обратном холодильнике должна подогреваться острым или глухим паром до 40–45°С, для того чтобы увлекаемый водяными парами р-нитротолуол не застыл там и не вызвал закупоривания. По окончании окисления выдавливают реакционную массу при 60–70°С на нучу. Последняя изготовляется из гомогенно освинцованного железа; для фильтрования служит кислотоупорная фильтровальная плитка. Горячие растворы тотчас спускают в эмалированные или же в гончарные сосуды. Из них в течение нескольких дней кристаллизуется большая часть взятого хрома в виде хромовокалиевых квасцов. Нитробензойную кислоту промывают на нуче до исчезновения серной кислоты, растворяют в разбавленном растворе соды, раствор фильтруют через мешок и осаждают технической соляной кислотой, отсасывают на нуче, промывают водой и высушивают при 40–50°С. Выход достигает самое меньшее 16 кг. Этиловый эфир р-нитробензойной кислоты. Для получения его берут тот же аппарат, как и для р-нитробензойной кислоты, только кроме обратного холодильника нужен еще и прямой холодильник. В аппарат загружают 16 кг сухой р-нитробензойной кислоты, заливают ее четырехкратным по весу количеством 96 %-ного этилового спирта, растворяют при помешивании, прибавляют 8 кг технической серной кислоты 66° Вё и нагревают без помешивания 16–20 часов с обратным холодильником до кипения. После охлаждения нейтрализуют при открытом люке медленно и осторожно водным раствором соды; для нейтрализации необходимо 11–12 кг соды Сольвэ. Отгоняют спирт с прямым холодильником, по охлаждении выдавливают остаток на глиняную нучу и промывают на последней эфир водой. Из фильтрата выделяют не вошедшую в реакцию нитробензойную кислоту технической соляной кислотой, отсасывают на нуче, промывают водой и высушивают. Выход 13,5–14,5 кг чистого эфира. Остаток получают в виде неизмененной кислоты обратно. Этиловый эфир р-аминобензойной кислоты. Чтобы получить и сырой продукт по возможности в чистом виде, раньше восстанавливали нитроэфир порошком олова и соляной кислотой. Полученную смесь после восстановления разбавляли большим количеством воды, точно нейтрализовали едким натром, нагревали до кипения и горячий водный раствор основания анестезина отделяли фильтрованием через небольшой фильтрпресс от выпавшего гидрата окиси олова. Технический анестезин, выпавший по охлаждении фильтрованного раствора, был достаточно чист. Маточники его можно было применять вместо чистой воды при новых операциях. Полученный таким образом технический продукт можно было превратить в чистое основание только одной перекристаллиацией из 80 %-ного спирта. Пасту гидрата окиси олова с фильтрпресса можно было продать, как таковую, или восстановить углем в металлическое олово в вагранках. Этот метод восстановления был бы хорош при условии, если бы он проводился в свободной от железа аппаратуре и технический продукт можно было очень легко очищать. Анестезин при соприкосновении с железом краснеет, так же как и калий сульфогваяколовый. Но финансовые убытки при продаже пасты окиси олова были значительны, а регенерация олова в вагранках для сравнительно небольших количеств была сложна. Поэтому в настоящее время повсюду восстановительный процесс ведут железом и уксусной кислотой и полученный таким образом технический продукт перед кристаллизацией перегоняют в вакууме. Анестезин можно перегонять уже при обыкновенном давлении при 305°С, но при этом к концу операции невозможно избежать разложения. Напротив, при остаточном давлении в 10–20 мм получается почти белый, слегка желтоватый продукт. Для восстановления железом и уксусной кислотой применяют аппарат, для которого достаточно объема в 200 л. Загружают в него 20 кг обезжиренных и просеянных чугунных опилок и 10 л воды. и травят при 70°, при помешивании 4 кг 80%-ной уксусной кислоты. Затем останавливают приток пара и при непрерывном помешивании постепенно приливают в аппарат 16 кг этилового эфира р-нитробензойной кислоты. Реакция сильно экзотермична, и вода выпаривается, хотя аппарат не подогревают. Время от времени приливают свежей воды взамен испаряющейся. По прилитии всего эфира, что берет 1½ – 2 часа, продолжают помешивание при легком подогревании еще 2 часа. Восстановление закончено, когда капли жидкости от восстановления более не окрашивают пропитанной раствором соды фильтровальной бумаги. Тогда дают жидкости остыть и нейтрализуют на лакмус раствором соды. Выделившееся при этом в свободном состоянии основание анестезина извлекают при 60° три раза, каждый раз 50 кг бензола. Последнюю вытяжку сохраняют, чтобы употребить вместо свежего бензола для первого извлечения следующей порции. Из двух первых бензольных вытяжек, высушенных хлористым кальцием, отгоняют бензол и перегоняют остаток – сильно окрашенное основание технического анестезина, – в вакууме. Служащий для этого маленький аппарат лучше всего делать из чистого алюминия; для наблюдения за перегонкой служит надставка из стекла или лучше из плавленого горного хрусталя. Прерывают перегонку, когда погон не стекает больше бесцветным. Нагревание можно производить маленькой газовой печью. Погон перекристаллизовывают из 80%-ного спирта. Для превращении основания, которое для этого должно быть чисто белым, в его хлоргидрат, растворяют его на холоду в абсолютном спирте и осторожно и медленно нейтрализуют раствор на конго 30%-ной спиртовой соляной кислотой. На следующий день выделившийся солянокислый анестезин отсасывают на нуче, промывают небольшим количеством холодного абсолютного спирта и высушивают при З0 – 40°. Выход по способу восстановления железом и уксусной кислотой бывает не менее 80% теории. 5.2.2.2 Синтез анестезина из р-толуидина Другим исходным продуктом для анестезина является р-толуидин. Он переводится в ацет-р-толуидин, а последний окислением марганцовокислым калием – в р-ацетаминобензойную кислоту. Эта кислота с одновременным образованием уксусного эфира переходит в этиловый эфир р-аминобензойной кислоты:  Ацетилирование. В колбе Шотта емкостью 15 л нагревают на масляной бане 5 кг р-толуидина и 6,5 кг 80%-ной уксусной кислоты в течение 12 часов до 140°. На колбу устанавливают широкую восходящую трубку, к которой присоединяют прямой холодильник. Нагревают очень медленно. Избыток уксусной кислоты отгоняется. Если последние часы держать температуру 135 – 140°, то ацетилирование оканчивается в 12 час. Содержимое колбы выливают в 25 л воды, выделившийся технический ацетат отсасывают на нуче и высушивают. Выход: 5,5 кг ацет-р-толуидина. Окисление. В деревянном чану емкостью 600 л с мешалкой и паровым змеевиком нагревают 500 л воды и 11 кг мелко порошкованного ацет-р-аминотолуидина до 30° и к раствору прибавляют сначала 8 кг мелко истолченного марганцевокислого калия. Температура сама поднимается до 40°. Небольшим подогреванием паром ее поднимают до 45°. Через 2 часа прибавляют еще 8 кг марганцевокислого калия и через 3 следующие часа – третью порцию, тоже 8 кг, так что всего уходит 24 кг марганцевокислого калия. После того как весь марганцевокислый калий прибавлен, перемешивают еще некоторое время, затем останавливают мешалку и дают осесть осадку перекиси марганца. Прозрачный раствор сливают, осадок переносят в мешки и отмывают его там от ацет-р-аминобензойной кислоты. Наконец осадок еще фугуют. Все растворы соединяют, и еще раз фильтруют. Из фильтрата осаждают прибавлением разбавленной серной кислоты до кислой реакции на конго, фильтруют, промывают и высушивают. Выход: 13 кг ацет-р-аминобензойной кислоты. Отщепление ацетильной группы и одновременное образование эфира. В колбе Шотта емкостью 15 л кипятят 1 час с обратным холодильником 3 кг ацет-р-аминобензойной кислоты с 6,8 кг 96%-ного спирта и 2,4 кг серной кислоты 66° Вё. После этого все выливают в 40 л воды и постепенно доводят поташом до щелочной реакции на фенолфталеин. Технический эфир отсасывают на нуче и высушивают. Он имеет желтоватый оттенок, который, если продукт не хотят перегонять в вакууме, можно удалить только тем, что его растворяют в десятикратном количестве воды с помощью свободной от железа соляной кислоты с прибавкой небольшого количества сернистой кислоты и угля для обесцвечивания, свободного от металла, и раствор фильтуют. Фильтрат снова осаждают поташом. Этот процесс растворения и осаждения иногда приходится повторять несколько раз. Наконец, предварительно очищенный таким образом эфир перекристаллизовывают из разбавленного спирта. Растворяют 5 кг технического эфира в 7 л 50%-ного спирта. Выкристаллизовывается 85 – 90 % эфира. Маточники можно использовать 1 – 2 раза для перекристаллизации, после чего из них отгоняют спирт. Из водного остатка выделяется технический эфир, который после промывания высушивают и снова перекристаллизовывают. Выход: 2,5 кг анестезина. 5.2.2.3 Изучен метод получения анестезина гидрированием этилового эфира n-нитробензойной кислоты на палладиевых катализаторах [22]. 5.2.4 Синтез бромкамфоры  Производство монобромкамфоры рентабельно потому, что в виде побочного продукта получается бромистоводородная кислота, имеющая большой спрос и потому довольно дорогая [21]. Аппаратура для бромирования аналогична применяемой при производстве трибромфенола.  Рис. 5.3 Аппаратура для бромирования трибромфенола. Так как загрузки делают едва ли больше 10 кг, то достаточна глиняная турилла емкостью 50 л и соответствующий глиняный горшок для улавливания бромистого водорода. Глиняную туриллу хорошо заменить фарфоровой и нагревать нужно не горячей водой, а на масляной бане, обогреваемой паром. Исходным продуктом является чистая естественная камфора, а не синтетическая. Последняя, даже если она относительно чиста, всегда дает худшие выхода, чем естественная камфора. В фарфоровую туриллу вносят 10 кг порошкованной камфоры и при частом помешивании толстой стеклянной палкой приливают в течение 3 часов точно 10,45 кг брома. Вследствие медленного приливания брома его можно производить из маленькой делительной воронки. Затем смесь нагревают в течение 3 часов до 80°С, поднимая после этого температуру в продолжение 3 часов до 100°С. Выделяющийся бромистый водород улавливается дистиллированной водой. По окончании выделения бромистого водорода выдавливают плав из фарфоровой туриллы в глиняный горшок, в который налит предварительно профильтрованный 5%-ный раствор соды. Помешивают до тех пор, пока плавающая на поверхности техническая бромкамфора не застынет в крупитчатую массу. Ее отсасывают на нуче и промывают холодной дистиллированной водой, пока промывные воды не перестанут реагировать с азотнокислым серебром. Водные растворы идут на приготовление бромистого натрия; техническую бромкамфору сушат при 35°С. Растворяют 3 части бромкамфоры в 4 части горячего 95 %-ного спирта, прибавляют к горячему раствору немного свободного от металлов обесцвечивающего угля и через 10 минут фильтруют через складчатый фильтр в большую колбу Эрленмейера для кристаллизации. Уже при обыкновенной температуре кристаллизуется около 50 % бромкамфоры в виде бесцветных иголок. Для усиления кристаллизации колбу ставят на несколько часов в смесь льда с солью, отсасывают кристаллы на нуче, споласкивают небольшим количеством спирта и сушат при 35°С. Маточник с промывным спиртом упаривают на водяной бане до ¼ объема и опять дают кристаллизоваться. Эту операцию повторяют еще раз. Последние довольно темные растворы от различных операций собирают и перерабатывают вместе. Их разбавляют полуторным по весу количеством дистиллированной воды, вследствие чего выделяется мазеобразное вещество. Его отделяют от прозрачного раствора, от которого отгоняют спирт. Из охлажденного остатка от дистилляции выделяется техническая бромкамфора, которую сушат и присоединяют к прочей технической бромкамфоре для очистки перекристаллизацией. Мазеобразное вещество сушат и определяют его точку плавления. Она обычно выше вследствие примеси непробромированной камфоры. Тогда продукт бромируют, беря 4–5 % от его веса брома, и опять определяют температуру плавления. Если она удовлетворительна, то продукт присоединяют к технической бромкамфоре. Если точка плавления этого вещества низка, то значит оно содержит дибромкамфору. Тогда его кипятят ½ часа с обратным холодильником с 10 % от его веса щелочи, выливают плав в воду, фильтруют, отмывают от щелочи и после сушки опять определяют температуру плавления. Выход: 122 % от взятой в работу камфоры, если это была чистая естественная камфора. Из синтетической камфоры выход тем ниже, чем меньше ее чистота. 5.2.4 Синтез валидола Ментиловый эфир валериановой кислоты можно приготовить пропусканием сухого хлористого водорода через смесь из моногидрата валериановой кислоты и ментола [21].  В достаточной величины круглодонную колбу из иенского стекла или пирекс, стоящую на паровой бане, вносят: 5,1 кг моногидрата валериановой кислоты и 7,8 кг ментола и пропускают в смесь сухой хлористый водород сначала 4 часа без нагрева, затем начинают постепенно нагревать, все время продолжая пропускать хлористый водород. Берут изредка пробы, взбалтывают с водой, нейтрализуют содой и смотрят, насколько прошел процесс этерификации. Когда он окончен (примерно через 7 часов пропускания хлористого водорода), смесь промывают в одной или нескольких склянках с тубусом у дна. Сначала основательно промывают водой, пока промывная вода почти не перестанет давать реакции с азотнокислым серебром. Затем следует промывка очень разбавленным раствором едкого натра (но не содой, так как вследствие выделения углекислоты в склянках может создаться избыточное давление, что опасно и может привести к потерям!). Сначала взбалтывают ½ мин., самое большее 1 мин., с маленьким избытком раствора щелочи, затем быстро отделяют раствор и опять промывают водой. Разбавленная щелочь и даже вода действуют на эфир омыляюще, хотя и медленно. Взбалтывание и отделение обоих слоев должно поэтому производиться очень тщательно. Отмывка от кислоты и последующая сушка обезвоженной глауберовой солью должны производиться с самим эфиром, а не с его раствором в эфире, бензоле или каких-либо других органических растворителях, так как они могут изменить запах продукта в неблагоприятную сторону. Полученный эфир должен быть обязательно перегнан в высоком вакууме при 3–5 мм остаточного давления. Эта операция необходима, иначе нельзя быть уверенным, что он на свету не пожелтеет. Он кипит при указанном вакууме пи 165–170°С. При перегонке его нельзя нагревать голым огнем горелки или на песчаной бане, надо на масляной бане, во избежание местных перегревов, которые вредно отражаются на запахе. После перегонки в вакууме определяют число омыления Кетсторфа. Для чистого продукта это число равняется 233. Нужно иметь в виду, что полное омыление продолжается очень долго – около 4 часов. Продажный Mentholum valerianicum имеет число омыления 162, так как содержит до 30 % свободного ментола. Посредством растворения в нем ментола продукт устанавливают на это число омыления, для чего ментола обычно идет 25–30 % от веса эфира. Выход из указанной загрузки: чистого эфира 11–11,5 кг, и продажного Mentholum valerianicum 14–14,5 кг. Чтобы продукт на свету оставался бесцветным не обладал чисты запахом, все исходные материалы должны быть возможно чище. Ментол должен быть фармакопейного качества. Чистая валериановая кислота очень дорога, поэтому берут техническую валериановую кислоту и очищают сами. Для очистки ее перегоняют из большой кварцевой или стеклянной реторты через колонку, наполненную стеклянными бусами или кольцами Рашига, прибавляя в колбу, смотря по степени препарата чистоты (предварительная проба) 3–6 % от ее веса бихромата натрия и соответствующее количество серной кислоты. Иногда техническая валериановая кислота настолько загрязнена (т. е. содержит смолистые вещества), что ее сначала приходится просто перегонять без колонки, а затем уже подвергать обработке хромпиком и серной кислотой и одновременно ректификации. Описанная очистка валериановой кислоты должна производиться на открытом воздухе под небольшой крышей, вследствие ее неприятного и чрезвычайно прилипчивого запаха. |