ОХТ(шпоры). УКлассификация технологических процессов механические и химические технологии промышленность неорганических веществ промышадность органических веществ (включая производство и переработку пищевых продуктов)
 Скачать 3.28 Mb. Скачать 3.28 Mb.
|

|
Технические условия, отраслевые, государственные международные стандарты. Примеры. Физико-химические закономерности технологических процессов. Классификация химических реакций: простые, сложные. Вероятность протекания химических реакций. Селективность процесса. Цепные и ступенчатые реакции. Цепные разветвленные и не разветвленные. Примеры. Способы регулирования скоростей и конверсии в цепных реакциях. Модели цепных процессов. Примеры. Ступенчатые реакции. Поликондесационное равновесие. Способы регулирования скоростей н конверсия ступенчатых реакций. Примеры. Кинетика химико - технологических процессов. Понятие о порядке реакций. Прямые и обратные реакций. Равновесие в технологических процессах. Примеры.
Процессы, протекающие в жидкостях и в газах, обычно рассматриваются с двух точек зрения процесс взаимодействия отдельных молекул на микроуровне и процесс взаимодействия агрегатов молекул - на макроуровне. В системе на микроуровне жидкость представляет собой свободные индивидуальные молекулы, движущиеся в различных направлениях, сталкивающиеся и смешивающиеся со всеми другими молекулами данной жидкости; в системе на макроуровне жидкость представляет собой совокупность большого числа небольших глобул, т. е. групп молекул, как бы заключенных в оболочку. Предполагается, что эта внешняя оболочка химически инертна и единственное назначение ее состоит в том, чтобы сохранить условно Снятую индивидуальность каждой глобулы. Весьма условно потоки жидкости в микро- и макро состоянии показаны на рис. Особенности микро- и макро состояния можно также проследить при смешении жидкостей: чем выше вязкость смешивающихся жидкостей, тем ярче проявляются в ней признаки макро состояния. В качестве примера представим себе сосуд с мешалкой, в котором весьма интенсивно перемешивается 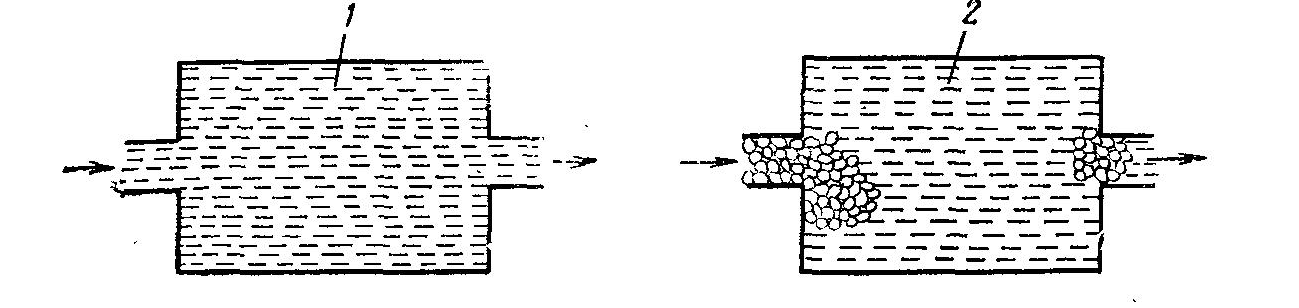 Поток идеализированных жидкостей в микро- и макро состояниях: 1 — жидкость в микро состоянии; 2—жидкость в макро состоянии. жидкость с малой вязкостью (вода). В эту жидкость вливают другую жидкость тоже малой вязкости (этиловый спирт). Эти жидкости хорошо перемешиваются и смесь быстро становится однородной; условно можно принять, что смешение происходит на микроуровне. Если в тот же сосуд вместо этилового спирта вливать очень вязкую жидкость (например, глицерин), а интенсивность перемешивания снизить (уменьшить частоту вращения мешалки), глицерин разобьется на отдельные капли (глобулы) и жидкость не скоро станет однородной. Условно можно принять, что смешение происходит на макроуровне. В микросостоянии жидкость не обладает никакими свойствами, обусловленными присутствием глобул молекул и, наоборот, свойства жидкости, находящейся в макросостоянии, в значительной степени определяются наличием глобул. Реальная смесь в той или иной степени проявляет промежуточные свойства, которые зависят от свойств жидкости и от системы, в которой происходит смешение. Все вопросы, изучаемые в курсе физической химии, химическое равновесие, кинетика химических реакций и др. рассматриваются на микроуровне, т. е. на уровне отдельных молекул. Такой подход позволяет проанализировать влияние различных факторов в идеализированных условиях. В промышленных условиях при оформлении подавляющего большинства химических процессов необходимо учитывать многие сопутствующие физические процессы, связанные с макро состоянием системы и накладывающиеся на основной химический процесс. Важнейшими из них являются: а) диффузия исходных реагентов в зону реакции и продуктов реакции из зоны реакции, б) выделение и распределение тепла. На оба эти процесса сильное влияние оказывают аэрогидродинамические условия (т. е. характер движения газа или жидкости), поскольку от них зависят конвективный перенос тепла и диффузия вещества.Таким образом, при изучении реального химико-технологического процесса необходимо учитывать влияние диффузии, теплопередачи и конвекции, т. е. процесс следует рассматривать на макроуровне.
На практике чаще всего используется классификация химических реакций по фазовому признаку, соответственно различают гомогенные и гетерогенные реакции. По этому же признаку наиболее часто подразделяют химические процессы и реакторы, в которых эти процессы осуществляются. В гомогенных системах все реагирующие вещества находятся водной какой-либо фазе: газовой (г), жидкой (ж) или твердой (т). В гетерогенных системах реагирующие вещества находятся в разных фазах: газ - жидкость (г - ж), газ - твердое (г - т), жидкость - твердое (ж - т), две несмешивающиеся жидкости (ж - ж) и две твердые фазы (т - т). Наиболее часто в промышленных процессах встречаются системы г - ж, г - т и ж - т. Иногда в промышленных процессах участвуют три или четыре фазы, например г - ж - т, г - ж - ж, г - ж - т - т. Обычно за отдельные фазы принимают только основные компоненты и не учитывают наличие малых количеств примесей. Так, например, в системах ж - ж и ж - т часто содержится газовая фаза, поскольку процессы проводятся в присутствии воздуха или других газов, или же в присутствии паров, так как жидкие компоненты частично испаряются. Но газовую фазу учитывают только в том случае, если она оказывает существенное влияние на процесс. Некоторые процессы начинаются в гомогенной среде, а затем в результате появления новой фазы система переходит в гетерогенную. Например, при получении полистирола к жидкому стиролу добавляют перекись бензоила и нагревают, при этом происходит полимеризация стирола с образованием новой фазы - твердого полистирола. Скорость реакции в гомогенных системах более высокая, чем в гетерогенных, так как в первом случае реакции протекают на уровне отдельных молекул (так называемый микроуровень). Поэтому в практических условиях обычно стремятся перевести гете рогенный процесс в гомогенный (путем плавления или растворения твердых реагирующих веществ, абсорбции или конденсации газов).
Строго гомогенные процессы, т. е. процессы, протекающие в одной фазе, встречаются в промышленности сравнительно редко, так как любое вещество содержит следы различных примесей, находящихся в другой фазе. Например, в 1 мл чистого горного возду содержится около 1000 взвешенных частиц, а в 1 мл дистиллированной воды до 20000 частиц. Поскольку следы инородных примесей часто активно влияют на ход процесса как катализаторы или ингибиторы, большинство процессов лишь условно можно отнести к гомогенным. Число таких условно гомогенных процессов велико в технологии и неорганических, и органических веществ. Например, окисление сероводорода и паров серы кислородом воздуха в производстве серной кислоты 2H2S + ЗО2 = 2S02 + 2Н2О + Q S + 02= 802 + Q протекает в гомогенной газовой фазе, несмотря на наличие в воздухе большого числа твердых частиц. К числу гомогенных процессов относят также окисление окиси азота до двуокиси азота кислородом воздуха в производстве азотной кислоты и многие другие. 2NO + О2 = 2N02 + Q Особенно многочисленны и разнообразны гомогенные процессы в газовой фазе, осуществляемые в технологии органических веществ. Примером этому может служить сжигание всевозможных видов газообразного топлива и, в частности, природного газа. Про цесс сжигания различного жидкого топлива также в большинстве случаев является гомогенным процессом, так как всякое жидкое топливо предварительно испаряется, а образовавшиеся пары затем окисляются кислородом воздуха. В технологии органических веществ сущность многих гомогенных процессов в газовой фазе состоит в том, что газообразные исходные вещества или пары, полученные испарением жидкости, обрабатываются тем или иным газообразным компонентом: хлором, сернистым ангидридом, окислами азота и др., при этом обычно протекают параллельные и последовательные реакции. Например, при термическом воздействии хлора на метан при 250-400 °С получают ряд соединений: СН4 + Сl2 = НСl + CH3Cl (хлористый метил) СН3С1 + Сl2 = НС1 + CH2Cl2 (хлористый метилен) СН2С12 + С12 = НС1 + СНСl3 (хлороформ) СНС13 + Cl2 = НС1 + ССl4 (четыреххлористый углерод). Из большого числа процессов, идущих в жидкой фазе, к гомогенным можно отнести процессы нейтрализации водных растворов кислот водными растворами щелочей. Например, при взаимодействии аммиачной воды и серной кислоты в коксохимическом производстве получают сульфат аммония 2NH4OH + H2S04 = (NH4)2S04 + 2Н20 К гомогенным реакциям относятся также некоторые обменные реакции, проходящие в растворах KCl + NaN03 = NaCl + KNO3 В жидкой фазе получают простые и смешанные эфиры из спиртов, например при разложении этилсульфата метиловым спиртом: C2H50S020H + СН3ОН = С2Н5ОСН3+ H2S04 В гомогенной среде осуществляют такой важный процесс, как получение адипиновой кислоты и многие другие. 3С6Н11ОН + 8HNO3 = 3С6Н10О4 + 7Н20 + 8NO Скорость гомогенных процессов Гомогенные процессы в большинстве случаев протекают в кинетической области, т. е. общая скорость процесса определяется скоростью химической реакции и подчиняется закономерностям, установленным для процессов, протекающих на микроуровне. Ско рости гомогенных процессов, в основе которых лежат простые необратимые реакции первого и второго порядков (A→R и 2A→R) выражаются уравнениями: dXA/dτn=1 = k(1-XA) dXA/dτn=2 = kCA,0(1-XA)2 из которых следует, что для повышения скорости необходимо увеличивать значения k (путем повышения температуры или применения катализатора) и СА,0. Так как на практике в большинстве случаев гомогенные реакции являются сложными (параллельными или последовательными), то в зависимости от значения констант скоростей этих реакций концентрация продуктов реакций будет изменяться во времени различно. При этом, чем выше порядок реакции, тем большее влияние оказывает концентрация исходного реагента на скорость соответствующей реакции. Например, при наличии двух параллельных реакций первого и второго порядков увеличение С А, 0 в два раза изменяет соотношение скоростей этих реакций в 4 раза. После интегрирования приведенных выше уравнений в пределах изменения ХА от 0 до ХА находим: τn=1 = 2,3/k*lg*(1-XA,0)/1-XA τn=2 = XA -XA,0/kCA,0(1-XA)*(1-XA,0) Если XA,0 = 0, то τn=1 = 2,3/k*lg*1/(1-XA,0) τn=2 = XA,0/kCA,0(1-XA) Полученные уравнения (а) - (г) позволяют определить время τ необходимое для достижения заданной конечной степени превращения ХА. Из этих уравнений следует, что константа скорости реакции первого порядка измеряется в ч-1, а реакции второго порядка - в м3 • кмоль-1 • ч-1. Гомогенный процесс на макроуровне протекает в том случае, когда на химическую реакцию накладываются другие физические или физико-химические процессы. Например, при взаимодействии двух жидких исходных реагентов или их растворов скорость гомо генного процесса зависит от условий и скорости перемешивания жидкостей; если химическое взаимодействие протекает при подогреве, то скорость гомогенного процесса будет зависеть также от способа подвода тепла.
Большинство химико-технологических процессов относятся к гетерогенным; при этом огромное разнообразие гетерогенных процессов затрудняет их классификацию. Механизм гетерогенных процессов сложнее гомогенных, так как реагирующие вещества находятся в разных фазах и подвод их к поверхности раздела фаз, где происходит химическое взаимодействие, а также массообмен между фазами осуществляются в результате молекулярной и конвективной диффузии, которые накладываются на основной хими ческий процесс. Усложнение вносят также процессы теплообмена и процессы, обусловленные особенностями гидродинамики потока. Только с учетом всех факторов, влияющих на технологический процесс, можно установить условия, обеспечивающие макси мальную его интенсивность, и управлять этим процессом. Примером гетерогенного процесса может служить процесс горения угля, который складывается из пяти стадий: внешняя диффузия 02 (через пограничный газовый слой); внутренняя диффузия 02 (через слой золы); химическая реакция; внутренняя диффузия СОг (через слой золы); внешняя диффузия СО2 (через пограничный слой газа). Горение угля сравнительно простой пример; на практике обычно протекают более сложные процессы. Однако в любом гетерогенном химическом процессе можно выделить три основных одновременно протекающих процесса:
Скорость гетерогенных процессов Для установления оптимальных параметров гетерогенных процессов и их аппаратурного оформления и проектирования необходимо прежде всего изучить статику (т. е. равновесие) и кинетику (т. е. скорость) этих процессов. Равновесие в гетерогенных системах зависит от температуры, давления и концентрации как исходных реагентов, так и продуктов реакции, скорость же взаимодействия реагентов, находящихся в разных фазах, зависит не только от скорости химической реакции, но и от многих других факторов (как любой процесс, протекающий на макроуровне). Поэтому в общем виде скорость гетерогенного процесса выражается следующим уравнением: r = KF ∆С где r - скорость гетерогенного процесса; К - коэффициент скорости процесса; F - поверхность контакта фаз; ∆С - движущая сила процесса. Коэффициент скорости процесса Коэффициент включает в себя многие факторы, влияющие на скорость гетерогенного процесса. В большинстве практических случаев влияние этих факторов неодинаково. Так, например, химический процесс обычно состоит из нескольких стадий, а его общая скорость определяется скоростью наиболее медленной (лимитирующей) стадии. Поэтому для интенсификации процесса необходимо прежде всего определить, какая из стадий является наиболее медленной, и ускорить ее. Такими наиболее медленными стадиями, каждая из которых может тормозить весь процесс, являются: 1 химическая реакция; 2 диффузия; 3 одновременно химическая реакция и диффузия. В первом случае, скорость диффузии велика по сравнению со скоростью химической реакции; тогда говорят, что процесс протекает в кинетической области. Во втором случае, скорость химической реакции значительно больше скорости диффузии - процесс протекает в диффузионной области (во внешне или внутридиффузионной). В третьем случае, скорости отдельных стадий соизмеримы, тогда говорят, что процесс протекает в переходной (смешанной) области. После установления лимитирующей стадии процесса принимают меры, обеспечивающие повышение скорости этой стадии. Так, если процесс протекает в кинетической области, создают условия, ускоряющие химическую реакцию; если процесс протекает в диф фузионной области, то ускоряют процесс диффузии; если же процесс протекает в переходной области, то создают условия, повышающие и скорость химической реакции, и скорость диффузии. Для установления лимитирующей стадии процесса существует не сколько приемов. Рассмотрим наиболее важные из них. Температура оказывает сильное влияние на скорость химических реакций. Так при увеличении температуры на 10°С скорость химической реакции в некоторых случаях возрастает в 2—3 раза. Скорость диффузии газов зависит от температуры в значительно меньшей степени, приближенно эта зависимость выражается уравнением: D = аТ2 где D - коэффициент диффузии; а - постоянный коэффициент. Из уравнения следует, что при повышении температуры на 100С скорость диффузии увеличивается всего на 3-5%. Это разное влияние температуры используют для определения лимитирующей стадии процесса. 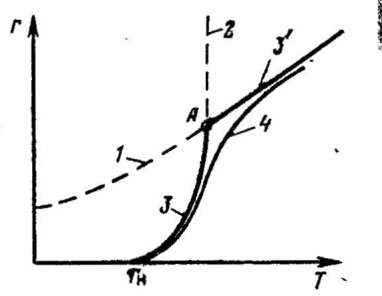 На рисунке кривая 1 отражает температурную зависимость скорости диффузии исходного реагента в зону реакции (rф). Кривая 2 отражает функциональную зависимость скорости химической реакции от температуры в соответствии с уравнением Аррениуса (rх.р.). Общая скорость отражена на рисунке двумя отрезками кривых 3 и 3'. Кривая 3 берет свое начало при температуре Тн, когда процесс химического взаимодействия начинает протекать с заметной скоростью. Затем наблюдается резкий подъем кривой. В точке А, где пересекаются кривые 1 и 2, скорость химической реакции и скорость диффузии равны rф = rх.р. = r. В дальнейшем величина r равна rф. Общая скорость процесса не может превышать самую низкую составляющую ее скорость rф ≥ r ≤ rх.р. Кривая 4 построена по опытным данным. Лимитирующая стадия гетерогенного процесса может быть уста новлена опытным путем. Так, если опыт показывает, что повышение температуры оказывает сильное влияние на скорость процесса, то процесс протекает в кинетической области (рис. III.3, область1). Если при дальнейшем повышении температуры ее влияние на скорость общего процесса уменьшается, значит процесс перешел в переходную область (рис. III.3, область2). Если же далее при повышении температуры общая скорость процесса практически не изменяется, значит процесс протекает в диффузионной области (рис. III. 3, область3).
Движущая сила процесса выражается уравнением ∆С = Сп.ф - Са.р. Где Сп.ф - концентрация исходного вещества в передающей фазе; Са.р - концентрация исходного вещества в той же фазе в зоне реакции. Из уравнения следует, что существуют два способа повышения ∆С: за счет увеличения Сп.ф и снижения Са.р. Движущая сила процесса возрастает, если увеличить концентрацию исходных реагентов в передающей фазе (Сп.ф.) или повысить давление, а так же если выводить продукты реакции из сферы взаимодействия. Увеличение давления является эффективным способом повышения ∆С, особенно для системы г - ж, так как в этом случае ∆С = рг - р* где рг - парциальное давление исходного газообразного реагента в газовой фазе; р* - равновесное давление исходного реагента у поверхности жидкости. При повышении давления процесса рг увеличивается, а р* остается постоянным, следовательно, при увеличении давления растет также ∆С 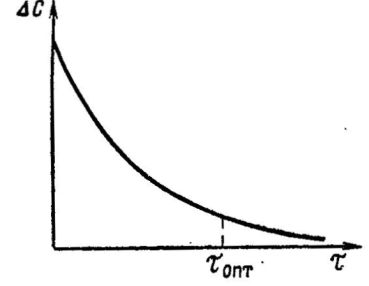 Вывод продуктов из сферы реакции также является эффективным способом увеличения движущей силы и широко используется в самых разнообразных производств венных процессах. При выводе продуктов реакции из зоны реакции уменьшается объем реакционной смеси, а Сг (или рг) соответственно увеличивается. Рост ∆С за счет снижения р* на практике осуществляется редко. Для систем г - ж это достигается путем уменьшения температуры так как одновременно понижается равновесное Давление вещества над жидкостью (р*). Однако практические возможности для использования этого приема ограничены, поскольку при понижении температуры сильно уменьшается константа скорости реакции (уравнение Аррениуса) и общая скорость процесса. По мере протекания реакции концентрация Сг уменьшается, соответственно снижается движущая сила ∆С, а также уменьшается общая скорость процесса, поэтому на практике в целях поддержания высокой скорости процесса его ведут при достаточно большом значении ∆С, ограничивая время пребывания реакционной смеси в реакторе некоторым оптимальным значением τопт.
Каталитические процессы в настоящее время составляют основу химической технологии, причем область их применения расширяется: около 90% новых производств, освоенных за последние годы химической промышленностью, основаны на взаимодействии, протекающем в присутствии катализаторов. Под катализом понимают изменение скорости химических реакций под воздействием веществ - катализаторов, которые, участвуя в процессе, остаются после его окончания химически неизменными. Катализ называется положительным, если катализатор ускоряет реакции, и отрицательным, если скорость реакции под воздействием катализатора снижается. Применение катализаторов облегчает практическое осуществление многих химических реакций; скорость некоторых из них увеличивается в тысячи и даже миллионы раз.. Очень многие промышленные процессы удалось осуществить только благодаря применению катализаторов. К числу каталитических процессов относятся важнейшие крупнотоннажные производства, например такие, как получение водорода, аммиака, серной и азотной кислот и многих других важнейших химических продуктов. Особенно велико и разнообразно применение катализа в технологии органических веществ и в производстве высокомолекулярных соединений. Отрицательный катализ применяется значительно реже: катализаторы, замедляющие скорость процесса называют также ингибиторами. Катализаторами могут быть вещества, находящиеся в любом яз трех агрегатных состояний. К твердым катализаторам можно отнести металлы и их оксиды, например Fe при синтезе аммиака, Pt при окислении аммиака, V2O5 при окислении S02, AI2O3 при крекинге нефтепродуктов и др. Жидкими катализаторами служат обычно кислоты и основания, например, H2S04 и Н3РО4 применяются при алкилировании ароматических углеводородов, при изомеризации н-бутилена в изобутилен и др. Примером газообразных катализаторов может служить BF3 в процессах полимеразиции некоторых углеводородов. Каталитические процессы можно разделить на две группы: гомогенные и гетерогенные. В гомогенно-каталитических реакциях реагирующие вещества и катализатор составляют одну фазу, а в гетерогенно-каталитических реакциях - разные фазы. В особую группу выделены микрогетерогенные и ферментативные каталитические процессы. Микрогетерогенный катализ происходит в жидкой фазе с участием коллоидных частиц металлов в качестве катализаторов. Ферментативный катализ наблюдается в биологических системах с участием сложных комплексов (часто белковой природы), называемых ферментами. Учение о катализе представляет очень интересную и увлекательную область знаний, имеющую исключительно большое практическое значение. В настоящее время в мире проводятся очень широкие и глубокие исследования в области изучения каталитических процессов; этим заняты десятки тысяч ученых.
Каталитические реакции подчиняются общим законам химии и термодинамики, но имеют при этом свои особенности, так как в них всегда участвует один дополнительный компонент - катализатор. Действие катализаторов принципиально отличается от действия других факторов, способствующих интенсификации химических реакций, например, температуры, давления, радиационного воздействия и др. Повышение температуры может ускорять реакцию вследствие увеличения энергетического уровня реагирующих молекул, т. е. их активации за счет вводимого извне тепла. При этом изменяется внутренняя энергия системы и смещается положение равновесия. Катализатор же не влияет ни на равновесие химической реакции, ни на все другие термодинамические характеристики реакций. Изменяя в равной степени скорость прямой и обратной реакций, катализатор способствует повышению скорости достижения равновесия при данных условиях. Теория каталитических процессов относится к числу сложных и недостаточно полно изученных областей современной физической Химии. В настоящее время еще нет общей теории, позволяющей предвидеть каталитическое действие различных веществ на ту или иную химическую реакцию. Существует несколько теорий, объясняющих механизм действия катализаторов, из которых наиболее распространенной теорией, служащей основой современных представлений о катализе, является теория промежуточных соединений. Согласно этой теории, медленную реакцию между исходными веществами можно заменить двумя или несколькими более быстрыми реакциями' с участием катализатора, который образует с исходными веществами промежуточные непрочные соединения. Ускоряющее действие катализатора состоит в понижении энергии активации реакций образующихся промежуточных соединений, что оказывает очень сильное влияние на скорость реакции, поскольку в уравнение Аррениуса: 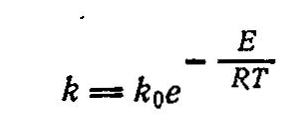 энергия активации Е входит в показатель степени. При практическом применении большое значение имеет технологическая характеристика промышленных катализаторов (активность, температура зажигания, производительность, селективность, отравляемость, прочность и др.). Наиболее важной характеристикой катализаторов является их активность, т. е. мера ускоряющего действия катализатора по отношению к данной реакции. Активность определяется уравнением: 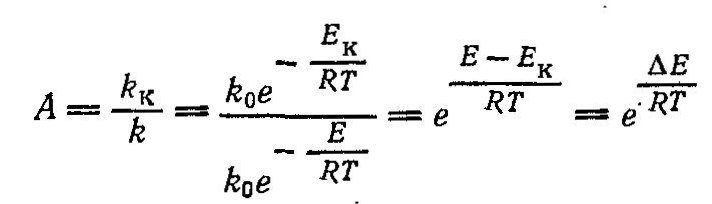 где А - активность катализатора; k, kk - константы скорости реакцнн без катализатора и в присутствии катализатора; ∆Е - снижение энергии активации под действием катализатора: Е, ЕК - энергия активации реакции без катализатора и в присутствии катализатора. Температурой зажигания катализатора называют минимальную температуру реагирующей смеси, при которой процесс начинает протекать с достаточной для практических целей скоростью. Чем активнее катализатор, тем ниже температура зажигания, что особенно важно при проведении экзотермических обратимых реакций, так как при этом соответственно повышается степень превращения. Отравление катализатора - это частичная или полная потеря его активности в результате действия посторонних примесей контактных ядов. Отравление может быть обратимым и необратимым. При обратимом отравлении примеси снижают активность катализатора временно, пока они присутствуют в зоне катализа; по удалении ядов катализатор восстанавливает свою прежнюю активность. При необратимом отравлении активность катализатора не восстанавливается и после удаления контактных ядов из зоны реакции. Активность твердого катализатора может снижаться также вследствие уменьшения активной поверхности катализатора под воздействием, например, высоких температур, при осаждении на поверхности катализатора продуктов реакции или пыли, механи ческого разрушения катализатора и по многим другим причинам. Важной особенностью катализаторов является их избирательность (селективность) по отношению к определенным реакциям. В сложных реакциях (параллельных и последова тельных), где термодинамически возможно образование нескольких продуктов, катализатор позволяет ускорить только одну целевую реакцию; естественно, что это имеет большое практическое значение. Для сложной реакции типа 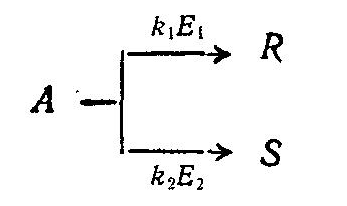 селективность выражается уравнением: 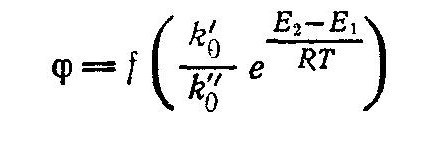 Из этого уравнения видно, что при некоторой заданной температуре Т путем подбора соответствующего катализатора можно изменять разность Е2 – Е1 и, таким образом, создавать возможности для получения только или главным образом целевого продукта. Особенно сильно селективность проявляется в сложных органических реакциях. Следовательно, применяя соответствующий катализатор, из одного и того же сырья можно получить различные целевые продукты.В присутствии катализатора обычно снижается порядок реакции, причем, чем активнее катализатор, тем этот эффект сказывается сильнее, т. е. тем ниже порядок реакции. В связи с этим, кинетика каталитических реакций обычно описывается уравнениями, найденными эмпирически, и формальный порядок таких каталитических реакций будет выражаться как в виде целого, так и дробного числа. В гомогенно-каталитических реакциях скорость процесса зависит от концентрации не только реагирующих веществ, но и катализатора. Основным недостатком гомогенного катализа является трудность выделения катализатора из конечной продукционной смеси, в результате чего часть катализатора теряется, а целевой продукт загрязняется. Однако в последнее время ведутся обширные иссле дования в области высокоактивных катализаторов гомогенного катализа, которые, присутствуя в малых дозах, вызывают цепные реакции. Поскольку количество вводимого катализатора невелико, после реакции он не извлекается из реакционной смеси, а остается в целевом продукте, не снижая качества получаемого целевого продукта. Большой интерес к гомогенному катализу объясняется главным образом тем, что при подборе соответствующих катализаторов интенсивность гомогенных процессов очень высока. Это объясняется тем, что гомогенные реакции протекают на микроуровне (на уровне отдельных молекул), когда вероятность столкновения молекул реагирующих веществ с молекулами катализатора весьма значительная.
Большинство известных промышленных каталитических реакций - это реакции между газообразными реагентами на твердых катализаторах. Изменение реакционного пути происходит в этом случае благодаря образованию промежуточных непрочных продуктов взаимодействия реагирующих веществ с катализатором. В общем случае процесс гетерогенного катализа на твердых пористых катализаторах складывается из нескольких элементарных стадий (рис): 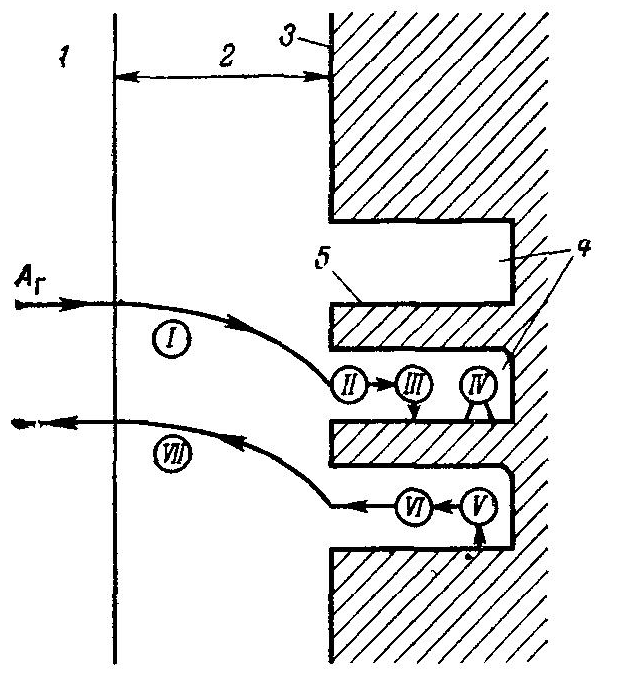  1 внешняя диффузия реагирующих веществ из ядра потока к поверхности зерен катализатора (через пограничный слой газа); 2 внутренняя диффузия реагентов в порах зерна катализатора; 3 активированная адсорбция веществ на поверхности катализатора с образованием поверхностных непрочных химических соединений активированных комплексов; 4 перегруппировка атомов с образованием поверхностных комплексов продукт - катализатор; 5 десорбция продукта с поверхности; 6 внутренняя диффузия продукта в порах зерна катализатора; 7 внешняя диффузия продукта реакции от поверхности зерна катализатора в ядро потока (через пограничный слой газа). В отличие от некаталитического гетерогенного процесса, протекающего в системе г-т, в данном случае появляются дополнительные промежуточные стадии, в частности, активированная адсорбция молекул исходных веществ на поверхности катализатора (стадия III). При этом желательно, чтобы твердые катализаторы имели большую легко доступную поверхность, что достигается уменьшением размера зерен и увеличением их пористости. В ряде случаев внутренняя поверхность таких катализаторов достигает десятков и даже сотен квадратных метров на 1 см3 катализатора. При наличии пористого катализатора реакция протекает как на внешней, так и на внутренней поверхности гранул катализатора. Часто внутренняя поверхность в тысячи раз превышает внешнюю поверхность, в этом случае влияние последней на процесс невелико. Так как при гетерогенном катализе процесс протекает главным образом на внутренней поверхности катализатора, то для описания большинства каталитических процессов более подходит квазигомогенная модель, а не модель частицы с невзаимодействующим ядром В настоящее время установлено, что не вся поверхность катализатора однородна, поэтому катализ осуществляется только на так называемых активных центрах.
В общем случае, суммарное уравнение скорости всего процесса гетерогенного катализа должно включать описание каждой из его стадий. Но точно так же, как и при протекании гетерогенного некаталитического процесса, не все его стадии оказывают равное влияние на скорость катализа. В большинстве случаев одна из стадий является наиболее медленной, лимитирующей процесс; она и определяет его скорость, поэтому для изыскания путей интенсификации такого процесса важно прежде всего установить лимитирующую стадию. Из анализа гетерогенных некаталитических процессов известно, что если наиболее медленной стадией, лимитирующей общую скорость, является диффузионный перенос газообразного вещества через пограничный слой газа, т. е., если процесс протекает во внешнедиффузионной области, эффективным средством его ускорения служит увеличение скорости газового потока. На этом основан наиболее часто применяемый экспериментальный метод определения влияния диффузии на скорость каталитического процесса. Для этой цели проводят серию опытов по определению скорости каталитической реакции при различной скорости потока реакционной смеси, но при постоянном отношении объема катализатора к объему смеси VK/Vr = const, или VK/fw = const (здесь VK — объем катализатора; Vr - объем реакционной газовой смеси; f - площадь сечения контактной трубки; w - линейная скорость газового потока). При увеличении w (что достигается уменьшением f) скорость реакции - rA = dXA/dτ будет возрастать только до тех пор, пока процесс протекает во внешнедиффузионной области (рис. IV. 3).  На участке кривой от w = 0 до w = w1 скорость потока оказывает влияние на скорость реакции и, следовательно, процесс протекает во внешнедиффузионной области. Влияние внутренней диффузии исследуют путем проведения серии опытов при скорости потока w > w1 (в области, где, внешняя диффузия уже не оказывает влияния на общую скорость процесса). Опыты проводят на зернах катализатора различного размера, результаты опытов выражают в виде графической зависимости Активное действие катализатора обусловлено прежде всего предварительной адсорбцией реагирующих веществ на поверхности катализатора, что оказывает большое влияние на скорость гетерогенного катализа. Адсорбция является самопроизвольным процессом, поэтому она сопровождается убылью энергии системы и связана с выделением тепла. Существует два вида адсорбции физическая и химическая (последняя называется также активированной адсорбцией, или хемосорбцией). Процесс катализа связан с хемосорбцией, но хемо- сорбция может быть обратимой и необратимой. Естественно, что в процессе катализа хемосорбция должна быть обратимой, так как активные центры должны непрерывно возобновлять свою функцио нальную деятельность по отношению к реагентам. Необратимая адсорбция вызывает отравление катализатора. С целью установления функциональной зависимости скорости гетерогенного катализа от различных факторов проводят всесторонние исследования, что позволяет определить влияние различных показателей на скорость адсорбции и десорбции реагентов поверх ностью катализатора, а также на скорость протекания других стадий, определяющих каталитический процесс в целом. Полученные при этом данные используют для составления теоретических уравнений, позволяющих установить общие закономерности в идеальных условиях. Для практических целей обычно применяют эмпирические уравнения, которые получают путем тщательного изучения влияния различных факторов на скорость конкретного каталитического процесса в условиях, близких к производственным. При этом идеа-лизированные модели служат отправной точкой для наиболее эффективного анализа экспериментальных данных. Нахождение кинетических уравнений и определение оптимальных параметров является главной целью научных исследований в области каталитических процессов, так как эти данные используются затем для расчета каталитических реакторов. Температура оказывает весьма существенное влияние на каталитические процессы, так как при повышении температуры увеличивается константа скорости реакции и одно временно изменяется константа равновесия. Для процессов, проходящих в кинетической области, повышение температуры всегда способствует приближению процесса к состоя нию равновесия. Но, как известно, для обратимых реакций равновесная степень превращения X* при повышении Т уменьшается для экзотермических реакций и увеличивается для эндотермиче ских реакций. Поэтому закономерности, отражающие суммарную скорость реакции и действительную степень превращения для экзотермических и эндотермических реакций, совершенно различны. При этом наблюдается такая же функциональная зависимость X = f(Т), как и для некаталитических процессов. Время контакта (время соприкосновения) реагирующих веществ с катализатором также является важной технологической характеристикой каталитического процесса, так как оно определяет его интенсивность. При расчете реакторов время контакта определяют по уравнению: τ = Vk/V где τ - время контакта; Vk - объем катализатора; V - объем реакционной смеси, проходящей через катализатор в единицу времени. Величина, обратная времени контакта, называется объемной скоростью и выражается уравнением S = 1/τ где S — объемная скорость (объем реакционной смеси, проходящей через еди ницу объема катализатора в единицу времени), м3 (газа) • м-3 (ката лиз.) • с-1 = с-1. При увеличении объемной скорости обычно снижается степень превращения, однако при этом возрастает интенсивность работы аппарата, т. е. увеличивается количество целевого продукта, получаемого с единицы объема катализатора в единицу времени. Это объясняется тем, что при увеличении скорости потока реагирующая система в большей мере удалена от равновесия, процесс протекает в области высоких скоростей за счет большой движущей силы ∆С = Рг - Рт. Интенсивность катализатора выражают в виде уравнения: G = pzS где G - производительность катализатора, кг/ч*м3 р - плотность реагента при нормальных условиях, кг/м3 z - мольная доля целевого продукта в газовой смеси; S - объемная скорость, ч-1. Из уравнения видно, что при увеличении объемной скорости производительность катализатора возрастает. Однако возможности для увеличения S ограничены, так как при этом степень превращения ХА уменьшается (снижается концентрация целевого продукта, что затрудняет выделение его из реакционной смеси), возрастает расход энергии и нарушается автотермичность экзотермической реакции вследствие относительного увеличения объема реакционной смеси. Применение давления является одним из способов повышения степени превращения при промышленном осуществлении обратимых каталитических реакций, проходящих с уменьшением объема. Давление становится решающим фактором, когда активность ка тализатора и величина X* низкие.
Промышленные твердые катализаторы обычно не являются индивидуальными веществами. Они представляют собой, за редким исключением, сложную смесь, называемую контактной массой. В контактной массе одни вещества являются собственно ка тализаторами, другие - носителями, а третьи служат активаторами. Носители - термостойкие, инертные, пористые вещества, на которые осаждением или другими способами наносят катализатор. Применение носителей улучшает свойства катализаторов и удешевляет их. В качестве носителей применяют пемзу, асбест, сили-кагель и другие пористые вещества. Активаторы или промоторы - вещества, повышающие активность основного катализатора, например, окислы щелочных металлов увеличивают активность железных катализаторов в синтезе аммиака и ванадиевых катализаторов при окислении двуокиси серы. Механизм действия активатора может быть самым различным: они могут образовывать химические соединения, твердые растворы, могут изменять электрофизические свойства поверхности и т. д. Активаторы могут также увеличивать активность катализатора, развивая и стабилизируя его поверхность. Последние называются структурными. Качество катализаторов характеризуют следующими основными показателями: активностью; избирательностью действия; устойчивостью к ядам и термостойкостью; механической прочностью; доступностью и дешевизной; теплопроводностью, которая должна быть возможно более высокой. Катализатор обычно готовят в виде зерен, таблеток, гранул. Иногда катализаторы применяют в виде тончайших сеток, изготовленных из металлов или сплавов. Наиболее часто катализаторы изготавливают следующим образом: 1 осаждением гидроокисей или карбонатов (из растворов их солей) на носителе с последующим формованием и прокаливанием; 2 совместным прессованием всех компонентов катализатора с вяжущим веществом; 3 сплавлением нескольких веществ; 4 пропиткой пористого носителя раствором, содержащим катализатор и активатор.
|