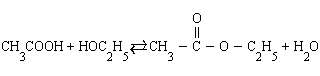6.Термохимия.Тепловые эффекты химических реакций.
Термохимия изучает тепловые эффекты химических реакций. Во многих случаях эти реакции протекают при постоянном объеме или постоянном давлении. Из первого закона термодинамики следует, что при этих условиях теплота является функцией состояния. При постоянном объеме теплота равна изменению внутренней энергии:
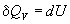 , , 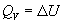 , (3.1) , (3.1)
а при постоянном давлении - изменению энтальпии:
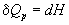 , , 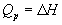 . (3.2) . (3.2)
Эти равенства в применении к химическим реакциям составляют суть закона Гесса:
Тепловой эффект химической реакции, протекающей при постоянном давлении или постоянном объеме, не зависит от пути реакции, а определяется только состоянием реагентов и продуктов реакции.
Другими словами, тепловой эффект химической реакции равен изменению функции состояния.
В термохимии, в отличие от других приложений термодинамики, теплота считается положительной, если она выделяется в окружающую среду, т.е. если 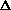 H < 0 или U < 0. Под тепловым эффектом химической реакции понимают значение H (которое называют просто "энтальпией реакции") или U реакции. H < 0 или U < 0. Под тепловым эффектом химической реакции понимают значение H (которое называют просто "энтальпией реакции") или U реакции.
Если реакция протекает в растворе или в твердой фазе, где изменение объема незначительно, то
H = U + (pV) 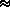 U. (3.3) U. (3.3)
Если же в реакции участвуют идеальные газы, то при постоянной температуре
H = U + (pV) = U + n . RT, (3.4)
где n - изменение числа молей газов в реакции.
Для того, чтобы облегчить сравнение энтальпий различных реакций, используют понятие "стандартного состояния". Стандартное состояние - это состояние чистого вещества при давлении 1 бар (= 105Па) и заданной температуре. Для газов - это гипотетическое состояние при давлении 1 бар, обладающее свойствами бесконечно разреженного газа.Энтальпию реакции между веществами, находящимися в стандартных состояниях при температуре T, обозначают 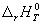 (r означает "reaction"). В термохимических уравнениях указывают не только формулы веществ, но и их агрегатные состояния или кристаллические модификации. (r означает "reaction"). В термохимических уравнениях указывают не только формулы веществ, но и их агрегатные состояния или кристаллические модификации.
Из закона Гесса вытекают важные следствия, которые позволяют рассчитывать энтальпии химических реакций.
Тепловой эффект - количество теплоты, выделившееся или поглощенное химической системой при протекании в ней химической реакции.
7.Закон гесса.
Пользуясь табличными значениями 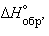 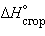 и и 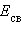 , можно рассчитать энтальпии различных химических процессов и фазовых превращений. Основанием для таких расчетов является закон Гесса, сформулированный петербургским профессором Г. И. Гессом (1841 г.): «Тепловой эффект (энтальпия) процесса зависит только от начального и конечного состояния и не зависит от пути перехода его из одного состояния в другое». , можно рассчитать энтальпии различных химических процессов и фазовых превращений. Основанием для таких расчетов является закон Гесса, сформулированный петербургским профессором Г. И. Гессом (1841 г.): «Тепловой эффект (энтальпия) процесса зависит только от начального и конечного состояния и не зависит от пути перехода его из одного состояния в другое».
Анализ закона Гесса позволяет сформулировать следующие следствия:
Энтальпия реакции равна разности сумм энтальпий образования конечных и начальных участников реакций с учетом их стехиометрических коэффициентов.
ΔH = ΣΔHобр.конечн – ΣΔHобр.нач
|
Энтальпия реакции равна разности сумм энтальпий сгорания начальных и конечных реагентов с учетом их стехиометрических коэффициентов.
ΔH = ΣΔHсгор.нач – ΣΔHсгор.конечн
|
Энтальпия реакции равна разности сумм энергий связей Eсв исходных и конечных реагентов с учетом их стехиометрических коэффициентов.
В ходе химической реакции энергия затрачивается на разрушение связей в исходных веществах (ΣEисх) и выделяется при образованиии продуктов реакции (–ΣEпрод). Отсюда
Следовательно, экзотермический эффект реакции свидетельствует о том, что образуются соединения с более прочными связями, чем исходные. В случае эндотермической реакции, наоборот, прочнее исходные вещества.
При определении энтальпии реакции по энергиям связей уравнение реакции пишут с помощью структурных формул для удобства определения числа и характера связей.
Энтальпия реакции образования вещества равна энтальпии реакции разложения его до исходных веществ с обратным знаком.
Энтальпия гидратации равна разности энтальпий растворения безводной соли 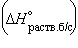 и кристаллогидрата и кристаллогидрата 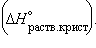
Из вышесказанного видно, что закон Гесса позволяет обращаться с термохимическими уравнениями как с алгебраическими, т. е. складывать и вычитать их, если термодинамические функции относятся к одинаковым условиям.
8.Самопроизвольные и не самопроизвольные процессы.второй закон термодинамики.
Второе начало термодинамики — физический принцип, накладывающий ограничение на направление процессов передачи тепла между телами.
Второе начало термодинамики гласит, что невозможен самопроизвольный переход тепла от тела, менее нагретого, к телу, более нагретому.
Второе начало термодинамики запрещает так называемые вечные двигатели второго рода, показывая что коэффициент полезного действияне может равняться единице, поскольку для кругового процесса температура холодильника не может равняться абсолютному нулю.
Второе начало термодинамики является постулатом, не доказываемым в рамках термодинамики. Оно было создано на основе обобщения опытных фактов и получило многочисленные экспериментальные подтверждения.
Существуют несколько эквивалентных формулировок второго начала термодинамики:
ПостулатКлаузиуса: «Невозможен процесс, единственным результатом которого являлась бы передача тепла от более холодного тела к более горячему»[1] (такой процесс называется процессом Клаузиуса).
ПостулатТомсона (Кельвина): «Невозможен круговой процесс, единственным результатом которого было бы производство работы за счет охлаждения теплового резервуара» (такой процесс называется процессом Томсона).
Эквивалентность этих формулировок легко показать. В самом деле, допустим, что постулат Клаузиуса неверен, то есть существует процесс, единственным результатом которого была бы передача тепла от более холодного тела к более горячему. Тогда возьмем два тела с различной температурой (нагреватель и холодильник) и проведем несколько циклов тепловой машины, забрав тепло Q1 у нагревателя, отдав Q2холодильнику и совершив при этом работу A = Q1 − Q2. После этого воспользуемся процессом Клаузиуса и вернем тепло Q2 от холодильника нагревателю. В результате получается, что мы совершили работу только за счет отъёма теплоты от нагревателя, то есть постулат Томсона тоже неверен.
С другой стороны, предположим, что неверен постулат Томсона. Тогда можно отнять часть тепла у более холодного тела и превратить в механическую работу. Эту работу можно превратить в тепло, например, с помощью трения, нагрев более горячее тело. Значит, из неверности постулата Томсона следует неверность постулата Клаузиуса.
Таким образом, постулаты Клаузиуса и Томсона эквивалентны.
Другая формулировка второго начала термодинамики основывается на понятии энтропии:
«Энтропия изолированной системы не может уменьшаться» (закон неубывания энтропии).
Такая формулировка основывается на представлении об энтропии как о функции состояния системы, что также должно быть постулировано.
Второе начало термодинамики в аксиоматической формулировке Рудольфа Юлиуса Клаузиуса (R. J. Clausius, 1865) имеет следующий вид[2]:
Для любой квазиравновесной термодинамической системы существует однозначная функция термодинамического состояния S = S(T,x,N), называемая энтропией, такая, что ее полный дифференциал dS = δQ / T.
9.Стандартная молярная энтропия простых и сложных веществ.
В химия, стандартная молярная энтропия будет энтропиясодержание одного моль вещества, под стандартными условиями (НЕ STP).
Стандартной молярной энтропии обычно дают символ So, и блоки j mol−1 K−1 (джоули в моль kelvin). Непохоже стандартные enthalpies образования, значение So абсолют. That is, элемент в своем стандартном положении имеет безнулевое значение So на температуре комнаты. Энтропия элемента может быть 0 j mol−1K−1 только на 0 k, согласно третий закон термодинамики. Однако, это предполагает что материал формирует «совершенный кристалл» без нисколько после того как он замер в энтропии (дефектах, вывихиваниях), которая никогда вполне поистине потому что кристаллы всегда растут на небесконечной температуре. Удачно эта остаточная энтропия часто довольно незначительна.
10.Критерии самопроизвольного протекание химических реакций.Энергия Гиббса.
Реальные процессы проводятся, как правило, в закрытых системах в изобарно-изотермических или изохорно-изотермических условиях. Критерием направленности самопроизвольного процесса в этих случаях является знак изменения энергии Гиббса DG или энергии Гельмгольца DА в системе.
G=H-TS=U+pV-TS, A=U-TS. TS хар-ет связанную с частицами системы энергию, т.е. ту часть полн. Е сист., кот. рассеив. в окр. среде в виде теплоты (потерянная работа). \энергия Гиббса (Гельмг.) – та часть полн. Е системы, кот. м.б. превр. в работу в изоб-изот Илии изох-изот проц. Е G и A – функции состояния системы, их абс. значения не поддаются вычислению. Для выяснения критерия направленности самопр. проц. в закр. сист: dG=dU+dpV+dVp-TdS-dTS, в силу TdS>=dU+pdV => dG<=-SdT+VdP. Для изобарно-изотермического: dG<=0, 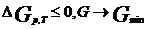 . В закрытой системе знак изменения энергии Гиббса является критерием направленности самопроизвольного процесса при проведении его в изоб.-изот. условиях. При DG=0 система нах. в сост. термод. равн. . В закрытой системе знак изменения энергии Гиббса является критерием направленности самопроизвольного процесса при проведении его в изоб.-изот. условиях. При DG=0 система нах. в сост. термод. равн.
Изохорно-изот. проц. в закр. сист. терм-ки возможен при DА<0, невозм, при DА>0, система нах. в термод. равн. при =0.
При const T, p=101,3 кПа: 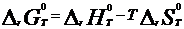 - Ур-ие Гиббса-Гельмгольца. При равенстве стандартной Е Гиббса нулю в системе устанавливается подвижное ТР, положение которого может смещаться в любую сторону при изменении внешних факторов. DG при 298 К можно рассчитать по стандартным Е Гиббса обр-я исходных веществ и продуктов реакции - Ур-ие Гиббса-Гельмгольца. При равенстве стандартной Е Гиббса нулю в системе устанавливается подвижное ТР, положение которого может смещаться в любую сторону при изменении внешних факторов. DG при 298 К можно рассчитать по стандартным Е Гиббса обр-я исходных веществ и продуктов реакции 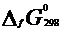 . Стандартной энергией Гиббса образования в-ва называют ст. Е Гиббса р-ции обр-я 1 моль данного соединения из простых в-в, находящихся в термод-ки устойчивых модификациях,кто провед. в станд. термод . услов. . Стандартной энергией Гиббса образования в-ва называют ст. Е Гиббса р-ции обр-я 1 моль данного соединения из простых в-в, находящихся в термод-ки устойчивых модификациях,кто провед. в станд. термод . услов.
Химический потенциал: для открытой системы G=f(p, T, ni), ni- кол-во в-ва i-го компонента, моль. Масса системы при этом const, но ее состав изменяется. Полный дифференциал : 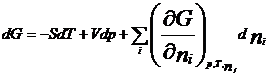 . Хим. потенциал i-го компонента: . Хим. потенциал i-го компонента: 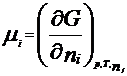 . По физ. смыслу : ХП означает изм. G сист. в PT процессе при доб. 1 моль i-го комп. к беск. большому кол-ву смеси, чтобы ее состав был const (nj=const). Для ид. р-ров: . По физ. смыслу : ХП означает изм. G сист. в PT процессе при доб. 1 моль i-го комп. к беск. большому кол-ву смеси, чтобы ее состав был const (nj=const). Для ид. р-ров: 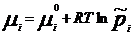 , , 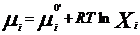 . . 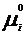 -стандартный хим. потенциал i-го комп. (при отн-ном давлении -стандартный хим. потенциал i-го комп. (при отн-ном давлении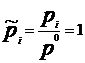 ), ХЭ и-ое – мольная доля I компонента. Второе уравнение справедливо для любых ид-х р-ров, первое – для газовых. Для индивид в-ва станд. хим. пот = станд. Е Гиббса обр-я в-ва. При проведении реакции в PT условиях: ), ХЭ и-ое – мольная доля I компонента. Второе уравнение справедливо для любых ид-х р-ров, первое – для газовых. Для индивид в-ва станд. хим. пот = станд. Е Гиббса обр-я в-ва. При проведении реакции в PT условиях: 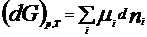 . Хим. переменная: . Хим. переменная: 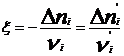 . (глубина, степень протекания реакции, число пробегов). . (глубина, степень протекания реакции, число пробегов). 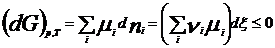 . Получим: . Получим: 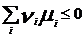 . – общие условия хим. равновесия и самопр. протек. реакции, идущей в закр. системе, при p, T=const. . – общие условия хим. равновесия и самопр. протек. реакции, идущей в закр. системе, при p, T=const.
11.Энтальпийный и энтропийный факторы химических реакций.
Энтальпийный и энтропийный факторы. Процессы могут протекать самопроизвольно (ΔG<0), если они сопровождаются уменьшением энтальпии (ΔH<0) и увеличением энтропии системы (ΔS>0). Если же энтальпия системы увеличивается (ΔH>0), а энтропия уменьшается (ΔS<0), то такой процесс протекать не может (ΔG>0). При иных знаках ΔS и ΔН принципиальная возможность протекания процесса определяется соотношением энтальпийного (ΔH) и энтропийного (ТΔS) факторов.
Если ΔН>0 и ΔS>0, т.е. энтальпийная составляющая противодействует, а энтропийная благоприятствует протеканию процесса, то реакция может протекать самопроизвольно за счет энтропийной составляющей, при условии, что |ΔH|<|TΔS|.
Если, энтальпийная составляющая благоприятствует, а энтропийная противодействует протеканию процесса, то реакция может протекать самопроизвольно за счет энтальпийной составляющей, при условии, что |ΔH|>|TΔS|.
12.Фазовые равновесия.
Равнове́сие фаз в термодинамике — состояние, при котором фазы в термодинамической системе находятся в состоянии теплового,механического и химического равновесия.
Типы фазовых равновесий:
Тепловое равновесие означает, что все фазы вещества в системе имеют одинаковую температуру.
Механическое равновесие означает равенство давлений по разные стороны границы раздела соприкасающихся фаз. Строго говоря, в реальных системах эти давления равны лишь приближенно, разность давлений создается поверхностным натяжением.
Химическое равновесие выражается в равенстве химических потенциалов всех фаз вещества.
Рассмотрим химически однородную систему (состоящую из частиц одного типа). Пусть в этой системе имеется граница раздела между фазами 1 и 2. Как было указано выше, для равновесия фаз требуется равенство температур и давлений на границе раздела фаз. Известно (см. статью Термодинамические потенциалы), что состояние термодинамического равновесия в системе с постоянными температурой и давлением соответствует точке минимума потенциала Гиббса.
Потенциал Гиббса такой системы будет равен
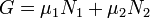 , ,
где μ1 и μ2 — химические потенциалы, а N1 и N2 — числа частиц в первой и второй фазах соответственно.
При этом сумма N = N1 + N2 (полное число частиц в системе) меняться не может, поэтому можно записать
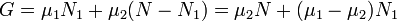 . .
Предположим, что 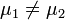 , для определенности, , для определенности, 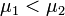 . Тогда, очевидно, минимум потенциала Гиббса достигается при . Тогда, очевидно, минимум потенциала Гиббса достигается при 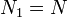 (все вещество перешло в первую фазу). (все вещество перешло в первую фазу).
Таким образом, равновесие фаз возможно только в том случае, когда химические потенциалы этих фаз по разные стороны границы раздела равны:
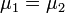 . .
13.Основные понятия химической кинетики.
Химическая кинетика или кинетика химических реакций — раздел физической химии, изучающий закономерности протекания химических реакций во времени, зависимости этих закономерностей от внешних условий, а также механизмы химических превращений.
14.Факторы влияющие на скорость химической реакции.
Скорость химической реакции зависит от природы реагирующих веществ и условий протекания реакции: концентрации с, температуры t , присутствия катализаторов, а также от некоторых других факторов (например, от давления - для газовых реакций, от измельчения - для твердых веществ, от радиоактивного облучения).
Влияние концентраций реагирующих веществ. Чтобы осуществля�лось химическое взаимодействие веществ А и В, их молекулы (части�цы) должны столкнуться. Чем больше столкновений, тем быстрее протекает реакция. Число же столкновений тем больше, чем выше концентрация реагирующих веществ. Отсюда на основе обширного экспериментального материала сформулирован основной за�кон химической кинетики, устанавливающий зависимость скорости реакции от концентрации реагирующих веществ:
Cкорость химической реакции пропорциональна произведению концентра�ций реагирующих веществ.
Для реакции ( I ) этот закон выразится уравнением
v = kcA cB , (1)
где сА и сВ - концентрации веществ А и В, моль/л; k - коэффициент пропорциональности, называемый константой скорости реакции. Основной закон химической кинетики часто называют законом действующих масс.
Из уравнения (1) нетрудно установить физический смысл константы скорости k : она численно равна скорости реакции, когда концентрации каждого из реагирующих веществ сос�тавляют 1 моль/л или когда их произведение равно единице.
Константа скорости реакции k зависит от природы реагирующих веществ и от температуры, но не зависит от их концентраций.
Уравнение (1), связывающее скорость реакции с концентрацией реагирующих веществ, называется кинетическим уравнением реакции. Если опытным путем определено кинетическое уравнение реакции, то с его помощью можно вычислять скорости при других концентрациях тех же реагирующих веществ.
Влияние температуры .
Зависимость скорости реакции от температу�ры определяетсяправилом Вант-Гоффа:
При повышении температуры на каждые 10о скорость большинства реакций увеличивается в 2-4 раза.
Математически эта зависимость выражается соотношением
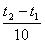
vt 2 = vt 1 γ ,
где vt 1 , vt 2 - скорости реакции соответственно при начальной ( t 1 ) и конечной ( t 2 ) температурах, а γ - температурный коэффициент скоро�сти реакции, который показывает, во сколько раз увеличивается ско�рость реакции с повышением температуры реагирующих веществ на 10°.
Правило Вант-Гоффа является приближенным и применимо лишь для ориентировочной оценки влияния температуры на скорость реак�ции. Температура влияет на скорость химической реакции, увеличивая константу скорости.
15.Энергетические диаграммы химических реакций.уравнение аррениуса.
Уравне́ние Арре́ниуса устанавливает зависимость константы скорости химической реакции 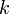 от температуры от температуры 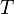 . .
Согласно простой модели столкновений химическая реакция между двумя исходными веществами может происходить только в результате столкновения молекул этих веществ. Но не каждое столкновение ведёт к химической реакции. Необходимо преодолеть определённый энергетический барьер, чтобы молекулы начали друг с другом реагировать. То есть молекулы должны обладать некой минимальной энергией (энергия активации 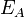 ), чтобы этот барьер преодолеть. Из распределения Больцмана для кинетической энергии молекул известно, что число молекул, обладающих энергией ), чтобы этот барьер преодолеть. Из распределения Больцмана для кинетической энергии молекул известно, что число молекул, обладающих энергией 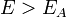 , пропорционально , пропорционально 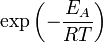 . В результате скорость химической реакции представляется уравнением, которое было получено шведским химиком Сванте Аррениусом из термодинамических соображений: . В результате скорость химической реакции представляется уравнением, которое было получено шведским химиком Сванте Аррениусом из термодинамических соображений:
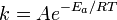
Здесь 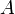 характеризует частоту столкновений реагирующих молекул, характеризует частоту столкновений реагирующих молекул, 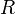 — универсальная газовая постоянная. — универсальная газовая постоянная.
В рамках теории активных соударений зависит от температуры, но эта зависимость достаточно медленная:
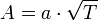
16.Химическое равновесие.
Химическое равновесие — состояние химической системы, в котором обратимо протекает одна или несколько химических реакций, причём скорости в каждой паре прямая-обратная реакция равны между собой. Для системы, находящейся в химическом равновесии, концентрации реагентов, температура и другие параметры системы не изменяются со временем.[1]
А2 + В2 ⇄ 2AB
17.Константа химического равновесия.
Конста́нта равнове́сия — величина, определяющая для данной химической реакции соотношение между термодинамическими активностями (либо, в зависимости от условий протекания реакции, парциальными давлениями, концентрациями или фугитивностями) исходных веществ и продуктов в состоянии химического равновесия (в соответствии с законом действующих масс). Зная константу равновесия реакции, можно рассчитать равновесный состав реагирующей смеси, предельный выход продуктов, определить направление протекания реакции.
Для реакции в смеси идеальных газов константа равновесия может быть выражена через равновесные парциальные давления компонентов pi по формуле[1]:
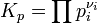
где νi — стехиометрический коэффициент (для исходных веществ принимается отрицательным, для продуктов — положительным). Kp не зависит от общего давления, от исходных количеств веществ или от того, какие участники реакции были взяты в качестве исходных, но зависит от температуры [2].
Например, для реакции окисления монооксида углерода:
2CO + O2 = 2CO2
константа равновесия может быть рассчитана по уравнению:
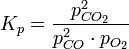
Если реакция протекает в идеальном растворе и концентрация компонентов выражена через молярность ci, константа равновесия принимает вид:
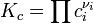
Для реакций в смеси реальных газов или в реальном растворе вместо парциального давления и концентрации используют соответственно фугитивность fi и активность ai:
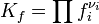
В некоторых случаях (в зависимости от способа выражения) константа равновесия может являться функцией не только температуры, но и давления. Так, для реакции в смеси идеальных газов парциальное давление компонента может быть выражено по закону Дальтона через суммарное давление и мольную долю компонента (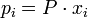 ), тогда легко показать[2], что: ), тогда легко показать[2], что:
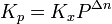
где Δn — изменение числа молей веществ в ходе реакции. Видно, что Kx зависит от давления. Если число молей продуктов реакции равно числу молей исходных веществ (Δn = 0), то Kp = Kx.
18.Зависимость константы хим равновесия от температуры.
Зависимость константы равновесия реакции от температуры может быть описана уравнением изобары химической реакции (изобары Вант-Гоффа):
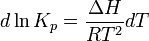
и изохоры химической реакции (изохоры Вант-Гоффа):
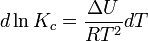
Здесь ΔH и ΔU — тепловой эффект реакции, протекающей, соответственно, при постоянном давлении или при постоянном объёме. Если ΔH > 0 (тепловой эффект положителен, реакция эндотермическая), то температурный коэффициент константы равновесия 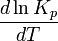 тоже положителен, то есть с ростом температуры константа равновесия эндотермической реакции увеличивается, равновесие сдвигается вправо (что вполне согласуется с принципом Ле Шателье). тоже положителен, то есть с ростом температуры константа равновесия эндотермической реакции увеличивается, равновесие сдвигается вправо (что вполне согласуется с принципом Ле Шателье). 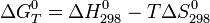
19.Принцип Ле Шателье.
При известных ΔH реакции или при Δn ≠ 0 на химическое равновесие можно воздействовать изменением температуры или давления. Химическое равновесие может быть смещено изменением концентраций реагентов. Другими словами, равновесие можно сместить внешним воздействием, руководствуясь принципомЛе Шателье: если на равновесную систему оказывать внешнее воздействие, то равновесие смещается в сторону, противодействующую этому воздействию.
Влияние температуры. Для реакций, идущих с уменьшением энтальпии (экзотермических), повышение температуры будет препятствовать протеканию прямого процесса, то есть смещать реакцию в сторону исходных веществ. Эндотермические реакции при этом будут смещаться в сторону конечных продуктов. Например, при обычных условиях реакция N2 + O2 не идет (ΔH > 0), но повышение температуры может сделать эти реакцию осуществимой. РеакцияCO + 1/2O2 = CO2, ΔH < 0 с повышением температуры будут смещаться в сторону исходных веществ.
Влияние давления. Если реагируют газообразные вещества, то при неизменном числе молей начальных и конечных реагентов повышение общего давления не приведет к смещению равновесия. Если число молей при реакции меняется, то изменение общего давления приведет к смещению равновесия. В частности, реакция 2CO + O2 = 2CO2, протекающая с уменьшением Δn, при повышении общего давления сместится в сторону образования СO2.
Влияние концентраций. В тех реакциях, в которых лучше оперировать концентрациями (реакции в растворах), увеличение концентраций исходных веществ приводит к смещению равновесия в сторону конечных продуктов и наоборот. Так, в реакции этерификации (образование сложного эфира)
-
увеличение концентрации уксусной кислоты или этанола увеличивает выход этилацетата, а добавление в систему воды приводит к омылению, т. е. образованию исходных продуктов.
20.Катализ.
Ката́лиз (греч. κατάλυσις восходит к καταλύειν — разрушение) — избирательное ускорение одного из возможных термодинамически разрешенных направлений химической реакции под действием катализатора(ов), который многократно вступает в промежуточное химическое взаимодействие с участниками реакции и восстанавливает свой химический состав после каждого цикла промежуточных химических взаимодействий.[1]
Катализ может быть положительным (когда скорость реакции увеличивается) и отрицательным (когда скорость реакции уменьшается). Для обозначения отрицательного катализа часто используют термин ингибирование.
Катализ бывает гомогенным и гетерогенным (контактным). В гомогенном катализе катализатор состоит в той же фазе, что и реактивы реакции, в то время, как гетерогенные катализаторы отличаются фазой.
Гомогенный катализ
Примером гомогенного катализа является разложение пероксида водорода в присутствии ионов йода. Реакция протекает в две стадии:
H2О2 + I → H2О + IO
H2О2 + IO → H2О + О2 + I
При гомогенном катализе действие катализатора связано с тем, что он вступает во взаимодействие с реагирующими веществами с образованием промежуточных соединений, это приводит к снижению энергии активации.
Гетерогенный катализ
При гетерогенном катализе ускорение процесса обычно происходит на поверхности твердого тела — катализатора, поэтому активность катализатора зависит от величины и свойств его поверхности. На практике катализатор обычно наносят на твердый пористый носитель.
Механизм гетерогенного катализа сложнее, чем у гомогенного. Механизм гетерогенного катализа включает пять стадий, причем все они обратимы.
Диффузия реагирующих веществ к поверхности твердого вещества
Физическая адсорбция на активных центрах поверхности твердого вещества реагирующих молекул и затем хемосорбция их
Химическая реакция между реагирующими молекулами
Десорбция продуктов с поверхности катализатора
Диффузия продукта с поверхности катализатора в общий поток
Примером гетерогенного катализа является окисление SO2 в SO3 на катализаторе V2O5 при производстве серной кислоты (контактный метод).
21. Энергетические диаграммы каталитических реакций
Скорость химической реакции при данной температуре определяется скоростью образования активированного комплекса, которая, в свою очередь, зависит от величины энергии активации. Во многих химических реакциях в структуру активированного комплекса могут входить вещества, стехиометрически не являющиеся реагентами; очевидно, что в этом случае изменяется и величина энергии активации процесса. В случае наличия нескольких переходных состояний реакция будет идти в основном по пути с наименьшим активационным барьером.
Катализ – явление изменения скорости химической реакции в присутствии веществ, состояние и количество которых после реакции остаются неизменными.
Различают положительный и отрицательный катализ (соответственно увеличение и уменьшение скорости реакции), хотя часто под термином "катализ" подразумевают только положительный катализ; отрицательный катализ называют ингибированием.
Вещество, входящее в структуру активированного комплекса, но стехиометрически не являющееся реагентом, называется катализатором. Для всех катализаторов характерны такие общие свойства, как специфичность и селективность действия.
Специфичность катализатора заключается в его способности ускорять только одну реакцию или группу однотипных реакций и не влиять на скорость других реакций. Так, например, многие переходные металлы (платина, медь, никель, железо и т.д.) являются катализаторами для процессов гидрирования; оксид алюминия катализирует реакции гидратации и т.д.
Селективность катализатора – способность ускорять одну из возможных при данных условиях параллельных реакций. Благодаря этому можно, применяя различные катализаторы, из одних и тех же исходных веществ получать различные продукты:
[Cu]: СО + Н2 ––> СН3ОН
|
[Al2О3]: С2Н5ОН ––> С2Н4 + Н2О
|
[Ni]: СО + Н2 ––> СН4 + Н2О
|
[Cu]: С2Н5ОН ––> СН3СНО + Н2
|
22.Типы растворов.
Раство́р — гомогенная (однородная) смесь, состоящая из частиц растворённого вещества, растворителя и продуктов их взаимодействия.
Раствор — однофазная система переменного состава, состоящая из двух или более компонентов. Растворы — гомогенные (однородные) системы, то есть каждый из компонентов распределён в массе другого в виде молекул, атомов или ионов[1].
Растворитель — компонент, агрегатное состояние которого не изменяется при образовании раствора. В случае же растворов, образующихся при смешении газа с газом, жидкости с жидкостью, твёрдого вещества с твёрдым, растворителем считается компонент, количество которого в растворе преобладает[1].
Образование того или иного типа раствора обусловливается интенсивностью межмолекулярного, межатомного, межионного или другого вида взаимодействия, то есть, теми же силами, которые определяют возникновение того или иного агрегатного состояния. Отличия: образование раствора зависит от характера и интенсивности взаимодействия частиц разных веществ[1].
По сравнению с индивидуальными веществами по структуре растворы сложнее[1].
Растворы бывают газовыми, жидкими и твёрдыми[1].
Твёрдые, жидкие, газообразные растворы
Чаще под раствором подразумевается жидкое вещество, например раствор соли или спирта в воде (или даже раствор золота в ртути — амальгама).
Существуют также растворы газов в жидкостях, газов в газах и жидкостей в жидкостях, в последнем случае растворителем считается вода, или же компонент, которого больше.
В химической практике обычно под растворами понимают гомогенные системы, растворитель может быть жидким, твёрдым (твёрдый раствор), газообразным. Однако нередко допускается и микрогетерогенность — см. «Золи».
«Раствором» именуют и смесь цемента с водой, песком и так далее. Хотя это и не является раствором в химическом смысле этого слова.
Ионные и коллоидные растворы
Коллоидные и ионные растворы (изучением коллоидных систем занимается коллоидная химия) отличаются главным образом размерами частиц.
В ионных растворах размер частиц менее 1×10−9 м, частицы в таких растворах невозможно обнаружить оптическими методами; в то время как в коллоидных растворах размер частиц 1×10−9 м — 5×10−7 м, частицы в таких растворах можно обнаружить при помощи ультрамикроскопа (см. эффект Тиндаля).
Растворение
Растворение — переход молекул вещества из одной фазы в другую (раствор, растворенное состояние). Происходит в результате взаимодействия атомов (молекул)растворителя и растворённого вещества и сопровождается увеличением энтропии при растворении твёрдых веществ и её уменьшением при растворении газов. При растворении межфазная граница исчезает, при этом многие физические свойства раствора (например, плотность, вязкость, иногда — цвет, и другие) меняются.
В случае химического взаимодействия растворителя и растворённого вещества сильно меняются и химические свойства — например, при растворении газа хлороводорода в воде образуется жидкая соляная кислота.
23.Объёмные и тепловые эффекты при растворении.
Тепловым эффектом химической реакции называется количество тепла, которое выделяется или поглощается при протекании реакции в условиях, когда исходные вещества и продукты реакции имеют одну и ту же температуру, система не производит никакой работы кроме работы против сил внешнего давления при постоянном объеме или давлении. Растворение большинства веществ также сопровождаются тепловыми эффектами.
В соответствии с первым началом термодинамики тепловой эффект реакции при постоянном объеме Qv равен приращению внутренней энергии системы ΔU, а тепловой эффект при постоянном давлении Qp равен приращению ΔH. Тепловой эффект считается положительными (термохимическая система знаков) или отрицательными (термодинамическая система знаков), если тепловая энергия в ходе реакции выделяется системой.
Раздел химической термодинамики, изучающий тепловые эффекты химических реакций, называется термохимией.
Если тепловой эффект прямым экспериментом определить нельзя, то его вычисляют, используя результаты вспомогательных калориметрических опытов. В основе таких расчетов лежит закон Гесса (1837 г.). В соответствии с этим законом тепловые эффекты химических реакций не зависят от пути, по которым протекает реакция, а зависят лишь от природы и физического состояния исходных веществ и продуктов реакции.
Например, при помощи прямого измерения трудно определить точно величину теплоты образования кристаллогидратов, так как реакция образования кристаллогидратов из безводного твердого вещества и воды идет быстро только вначале, пока не прореагируют с водой поверхностные слои кристалликов безводного вещества, а затем реакция сильно замедляется и долго не заканчивается. Кроме того, процесс осложняется растворением вещества в воде. Однако при помощи основного закона термохимии можно определить теплоту образования кристаллогидрата. Для этого надо определить теплоту растворения безводной соли и теплоту растворения кристаллогидрата, и из первой величины вычесть вторую:
ΔН = ΔНбезв - ΔНкрист
Теплота растворения вещества увеличивается с увеличением количества растворителя, приходящегося на 1 моль растворяемого вещества. Поэтому под тепловым эффектом растворения понимают количество выделившейся или поглощённой теплоты при растворении 1 моля вещества в определенном количестве (не менее 400 молей) растворителя. Дальнейшее разбавление раствора очень слабо изменяет величину теплоты растворения. В целом можно сказать, что теплотой растворения принято называть количество теплоты, поглощающейся или выделяющейся при растворении одного моля вещества в таком количестве растворителя, когда дальнейшее его добавление не сопровождается измеримым тепловым эффектом.
Отнесенное к одному молю растворенного вещества, полное изменение энтальпии при растворении nв-ва молей вещества в nраст-ля. молях растворителя называетсяинтегральной теплотой растворения и обозначается ΔHm. Индекс m - это численное значение концентрации раствора в молярной шкале: 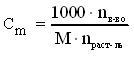 . Важное преимущество молярной шкалы перед другими концентрационными шкалами заключается в том, что при изменении концентрации растворенного вещества количество молей растворителя не меняется, то есть nраст-ля = const. . Важное преимущество молярной шкалы перед другими концентрационными шкалами заключается в том, что при изменении концентрации растворенного вещества количество молей растворителя не меняется, то есть nраст-ля = const.
Интегральная теплота растворения зависит от температуры и концентрации, поэтому указание этих характеристик процесса является обязательным. В литературе по термохимии концентрацию раствора обычно выражают величиной разбавления, то есть числом молей растворителя, приходящимся на 1 моль растворенного вещества, а количественное соотношение компонентов при растворении представляют термохимическим уравнением, например:
Особый интерес представляет первая интегральная теплота растворения - изменение энтальпии при растворении 1 моля вещества в бесконечно большом количестве растворителя. В результате процесса образуется бесконечно разбавленный раствор, например,
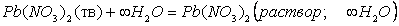
Растворение твёрдых веществ складывается из нескольких процессов, каждый из которых сопровождается тепловым эффектом:
разрушение кристаллической решётки, сопровождающееся эндотермическим эффектом (ΔНкр.реш. > 0);
процесс сольватации или гидратации, сопровождающееся экзотермическим эффектом (ΔНсольв. < 0);
процесс диффузии, но этот эффект настолько мал, что его в данном случае не учитывают.
Следовательно, теплота растворения твёрдого тела определяется алгебраической суммой двух теплот:
ΔНраст. = ΔНкр.реш. + ΔНсольв.
При сольватации (гидратации) всегда выделяется теплота, то есть ΔНсольв. < 0.
Величина же ΔНкр.реш. может быть отрицательной (при растворении газов) и положительной (при растворении твёрдых веществ).
Поэтому вещества, обладающие прочной кристаллической решеткой и слабо гидратирующиеся в растворе, растворяются с поглощением теплоты. Вещества же с непрочной кристаллической решеткой, образующие в растворе сильно гидратированные ионы, например гидроксид-ионы, растворяются с выделением теплоты.
Исследование теплот растворения и зависимости их от концентрации позволяет получить много информации о строении раствора.
24.Свойства растворов неэлектролитов.
|
 Скачать 237.79 Kb.
Скачать 237.79 Kb.