химия. 1 Предмет и методы химической термодинамики. Взаимосвязь между процессами обмена веществ и энергии в организме. Химическая термодинамика как теоретическая основа биоэнергетики. Термодинамика
 Скачать 0.58 Mb. Скачать 0.58 Mb.
|

|
Буферным действием обладают: -Система слабая кислота – ее соль с сильным основанием, а также сочетание кислой и средней солей слабых кислот или двух кислых солей; -Система слабое основание – его соль с сильной кислотой; -Ионы и молекулы амфолитов – аминокислотные и белковые системы. Основная функция буферных систем - предотвращение значительных сдвигов рН путём взаимодействия буфера, как с кислотой, так и с основанием. Действие буферных систем в организме направлено преимущественно на нейтрализацию образующихся кислот. В организме одновременно существует несколько различных буферных систем. В функциональном плане их можно разделить на бикарбонатную и небикарбонатную. Небикарбонатная буферная система включает гемоглобин, различные белки и фосфаты. Она наиболее активно действует в крови и внутри клеток. 7)Роль воды и растворов в жизнедеятельности. Физико-химические свойства воды, обусловливающие её уникальную роль, как единственного биорастворителя. Автопротолиз воды. Константа автопротолиза воды. Водородный показатель рН. Зависимость растворимости веществ в воде от соотношения гидрофильных и гидрофобных свойств; влияние внешних условий на растворимость. ТД растворения. Понятие об идеальном растворе. Растворимость газов в жидкостях и её зависимость от разных факторов: массовая доля, молярная и моляльная концентрация. Титр раствора. Многие химические процессы протекают только если участвующие в них вещества находятся в растворенном состоянии. Раствор - это термодинамически устойчивая гомогенная физико-химическая многокомпонентная система переменного состава. Учение о растворах важно для медиков, т.к.: Важнейшие биологические жидкости - кровь, лимфа, слюна и т.п. являются растворами солей, белков, углеводов, липидов в воде. Биохимические реакции в организмах протекают в растворах. Усвоение пищи происходит с переходом питательных веществ в растворенное состояние. Биологические жидкости транспортируют питательные вещества (жиры, аминокислоты), лекарственные препараты к органам и тканям, а также выводят метаболиты (мочевину, углекислый газ). Вода хорошо растворяет ионы и многие полярные соединения. Диэлектрическая проницаемость воды обуславливает это свойство (ε= 78,5). Притяжение между ионами уменьшается примерно в 80 раз при растворении в воде. Автопротолиз – обратимый процесс передачи протона от одной нейтральной молекулы жидкости к другой и образования в результате равного числа катионов и анионов. Н2О+H2O = (Н3О+) + (ОН-) К(H2O) - константа автопротолиза воды. Константа автопротолиза воды зависит от температуры. Произведение концентраций ионов водорода и гидроксид-ионов при 25°С есть величина постоянная, называемая ионным произведением воды (Кw): Kw = [H+] · [OH-] = 10^-14. Водородный показатель (рН) – величина, характеризующая активность или концентрацию ионов водорода в растворах. Водородный показатель обозначается рН. Водородный показатель численно равен отрицательному десятичному логарифму концентрации ионов водорода, выраженной в молях на литр: pH = -lg[H+]. С помощью водородного показателя кислотность водных растворов характеризуется следующим образом: pH = 7 – среда нейтральная pH < 7 – среда кислая pH > 7 – среда щелочная При растворении дифильных веществ происходит изменение структуры воды так как результат взаимодействия с гидрофобными группами сводится к минимуму. Гидрофобные группы, ассоциируясь, выталкивают молекулы воды из области своего расположения. Термодинамика процесса растворения ΔG=ΔH–TΔS < 0. ΔH – энтальпийный фактор, TΔS– энтропийный фактор. При растворении жидких и твёрдых веществ энтропия системы обычно возрастает (ΔS>0), т.к. растворяемые вещества из более упорядоченного переходят в менее упорядоченное. Вклад энтропийного фактора, особенно заметен при повышенных Т, т.к. произведение TΔS – велико, в результате возрастает убыль ΔG. При растворении газов ΔS<0, т.к. вещество из менее упорядоченного переходит в более упорядоченное, а снижение T способствует растворению газов. Зависимость растворимости газов в жидкости определяется законом Генри: Растворимость газа в жидкости прямо пропорциональна парциальному давлению газа при постоянной температуре. Условием равновесного распределения вещества между газом и жидкостью является равенство химических потенциалов между жидкой и газовой фазами: Mж=Мг C=Г·P – формульное выражение закон Генри, где: Г – постоянная Генри; C – мольная доля растворенного вещества; Р – парциальное давление газа. Уравнение применимо для идеальных растворов. Если происходит диссоциация или ассоциация: Cn=Г·P, где: n – коэффициент, учитывающий изменение числа частиц в растворе. Растворимость газа в жидкости зависит от ряда факторов: природа растворителя и растворимого вещества, давления, газовой фазы и температуры. Идеальный раствор – раствор, образование которого не сопровождается химическим взаимодействием, изменением объёма и тепловым эффектом. И. М. Сеченов, изучая растворимость газов, установил, что присутствие электролитов понижает растворимость газов:ln (C0/C) = kc · Сэл, где: С – искомая растворимость газа; С0 – растворимость газа в чистом растворителе; k – константа Сеченова Cэ – концентрация электролита Способы концентрации водных растворов: Массовая доля (массовая процентная концентрация ω%) – показывает кол-во грамм растворённого вещества содержится в 100 г раствора. ω% = m (в-ва) *100%/ m (р-ра). Мольная доля (N) показывает количество молей растворённого вещества в суммарном количестве молей раствора N = ν (в-ва)/ ν (р-ра). Моляльная концентрация Cm (моль/кг) показывает количество молей растворённого вещества в 1 кг растворителя. Сm = v (в-ва)/m (р-ля). Молярная концентрация CM (моль/л) показывает количество молей растворённого вещества в 1 л раствора. СM = v (в-ва)/V (р-ра). Титр (Т) показывает массу вещества в граммах, содержащаяся в 1 мл (1 см3) раствора. Размерность г/см3. Т=m/V 8)Основы титриметрического анализа: прямое, обратное, косвенное титрование. Химический эквивалент вещества, фактор эквивалентности. Молярная концентрация эквивалента. Закон эквивалентов. Точка эквивалентности и способы её фиксирования. Расчёт массовой доли вещества по данным титриметрического анализа. Использование титр.методов в медицине. Одним из наиболее распространенных и доступных методов аналитической химии является титриметрический, предложенный в 1880 году Гей-Люссаком. Титрование – процесс непрерывного добавления одного раствора (точно известной концентрации) к другому (концентрацию которого необходимо установить) до достижения точки эквивалентности (т.э.). Момент, при котором к титруемому компоненту добавлено эквивалентное количество титранта (раствора с известной концентрацией вещества), отвечающий стехиометрическому уравнению взаимодействия, называется точкой эквивалентности. Достоинства титриметрического анализа: Быстрота определения Простота оборудования Возможность автоматизации Точность определения – относительная погрешность 0,1-0,01%. Возможность определять как малые, так и большие количества определяемых веществ. Возможность определять два и более вещества одновременно. Ограничениями метода являются: Требуется стандартизация титранта. Стандартизация титранта – использование вспомогательного титрования, для определения точной концентрации титранта. Для р-ра HCl используют декагидрат карбоната Na. Для р-ра NaOH – дигидрат щавелевой кислоты. Невозможность использования нестехиометрических реакций, кинетических реакций. Невысокая чувствительность Титриметрические методы анализа служат для определения основных компонентов и примесей. Титрование применяют в анализе органических и неорганических соединений, а так же биологических жидкостей. Различают три основных способа титрования: прямое, обратное, косвенное или заместительное. При прямом титровании используют исследуемый и один рабочий растворы. В процессе определения к определённому точно измеренному объёму одного из них по каплям добавляют второй раствор до наступления точки эквивалентности. N1V1 = N2V2.При обратном титровании используют исследуемый и два рабочих раствора, один их которых является вспомогательным, а второй применяют для титрования. В процессе анализа к определённому точно измеренному объёму исследуемого раствора одномоментно добавляют взятый в избытке фиксированный объём вспомогательного рабочего раствора. В результате протекания химической реакции вещество, присутствующее в исследуемом растворе, расходуется полностью. Не прореагировавший избыток вещества вспомогательного раствора титруется затем вторым рабочим раствором до наступления точки эквивалентности. Обратное титрование в аналитической практике может называться иначе титрованием по остатку или с двумя титрантами. Оно используется, если определяемое вещество не реагирует или реагируют медленно с веществом второго рабочего раствора, либо в реакции между ними невозможно определить точку эквивалентности. N2V2 = N1V1 + N3V3.При косвенном, или заместительном, титровании также используют исследуемый раствор и два рабочих раствора. В ходе анализа к точно измеренному объёму исследуемого раствора одномоментно добавляют нефиксированный заведомый избыток первого рабочего раствора. В результате протекающей реакции вещество исследуемого раствора полностью расходуется с образованием эквивалентного количества соответствующего продукта реакции, который затем титруется вторым рабочим раствором до наступления точки эквивалентности. N1V1 = N2V2. Эквивалент – это реальная или условная частица, которая в кислотно-основных реакциях присоединяет (или отдает) один ион Н+ или ОН–, в окислительно-восстановительных реакциях принимает (или отдает) один электрон, реагирует с одним атомом водорода или с одним эквивалентом другого вещества. Число, показывающее, какая часть молекулы или другой частицы вещества соответствует эквиваленту, называется фактором эквивалентности (fЭ). Фактор эквивалентности – это безразмерная величина, которая меньше, либо равна 1. Молярная масса эквивалента вещества х. Это масса 1 моля эквивалента этого вещества, равная произведению фактора эквивалентности на молярную массу вещества х: Мэ = fэкв· М(х) Молярная концентрация эквивалента определяется числом моль-эквивалентов на 1 литр раствора. Эквивалент определяется в соответствии с типом рассматриваемой реакции. Сэ = nэ/V (моль-экв/л) Закон эквивалентов: Все вещества взаимодействуют друг с другом в количествах, пропорциональных их эквивалентам. Эквивалентное соотношение означает одинаковое число моль эквивалентов. Закон эквивалентов можно сформулировать иначе: число моль-эквивалентов для всех веществ, участвующих в реакции, одинаково. Способы фиксирования точки эквивалентности. В химических методах анализа различают: Безындикаторный. Один из растворов выступает в роли индикатора, например, в перманганатометрии, рабочий раствор - перманганат калия имеет интенсивную малиновую окраску. Индикаторный, который предполагает использование интенсивно окрашенных индикаторов. В инструментальных или физико-химических методах анализа точку эквивалентности определяют по изменению показания прибора (потенциометрическое, кулонометрическое, кондуктометрическое титрование) в ходе титрования. Формулы для расчетов в титриметрическом анализе: 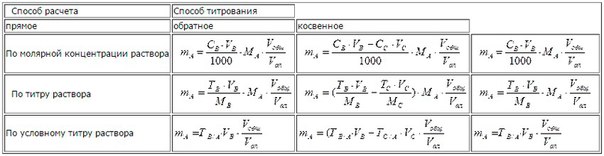 Где: mA – масса определяемого вещества, г; МА, МВ, МС – молярные массы веществ А, В, С соответственно (г/моль); CB, TB, VB – молярная концентрация (моль/л), титр (г/мл) и объем (мл) стандартного раствора В; CC, TC, VC – то же для стандартного раствора С; ТВ/А – титр раствора В по веществу А (г/мл); ТС/А – титр раствора С по веществу А (г/мл); Vобщ - общий объем анализируемого раствора, мл; Vал – объем аликвоты. Количественный анализ является одним из разделов аналитической химии, который имеет важное значение в современной теоретической и практической медицине. Практическому врачу необходимы данные количественного состава биологических жидкостей, чтобы судить об их нормальном или патологическом состоянии. Так, определение в плазме крови ионов Na+ и K+ имеет важное значение, поскольку нормальный ритм сердца во многом зависит от соотношения их концентраций. 9)Коллигативные свойства разбавленных растворов неэлектролитов и электролитов. Закон Рауля и следствия из него: понижение температуры кипения растворов, осмос. Осмотическое давление, закон Вант-Гоффа. Коллигативные свойства – это свойства растворов, зависящие от числа частиц растворенного вещества. К их числу относятся: Понижение давления паров Повышение температуры кипения Понижение температуры затвердевания (кристаллизации) Осмотическое давление раствора. Понижение давления паров Давление насыщенного пара (т.е. пара, который пребывает в состоянии равновесия с жидкостью) над чистым растворителем называется давлением или упругостью насыщенного пара чистого растворителя. Если в некотором растворителе растворить нелетучее вещество, то равновесное давление паров растворителя при этом понижается, т.к. присутствие какого-либо вещества, растворенного в этом растворителе, затрудняет переход частиц растворителя в паровую фазу. Экспериментально доказано, что такое понижение давления паров напрямую зависит от количества растворенного вещества. В 1887 г. Ф.М. Рауль описал количественные закономерности коллигативных свойств растворов. Первый закон Рауля: По свойствам растворы отличаются от чистых растворителей, например, давление пара растворителя над раствором ниже, чем над чистым растворителем. Это понижение прямо пропорционально мольной доле растворённого нелетучего неэлектролита (закон Рауля): Δp = К · χ, где: Δp — понижение давление пара над раствором по сравнению с чистым растворителем; χ — мольная доля растворённого вещества; К — коэффициент пропорциональности, равен Δp при χ=1 Второй закон Рауля: Также Рауль экспериментально доказал, что повышение температуры кипения раствора по сравнению с температурой кипения растворителя, а равно и понижение температуры замерзания раствора по сравнению с аналогичным характеризующей величиной для растворителя прямо пропорциональна моляльности раствора, то есть, ΔTкип/зам = Kэб/кр · Сmв-ва, где: Kэб/кр — соответственно эбулиоскопическая (+0,52 у воды) (от лат. ebullire — «кипеть» и др.-греч. σκοπέω — «наблюдаю») и криоскопическая (–1,86 у воды) (относится к замерзанию) константы, характерные для данного растворителя; Сmв-ва — моляльность вещества в растворе. Растворы электролитов не подчиняются Законам Рауля!!! Но для учёта всех несоответствий Вант-Гофф предложил ввести в приведённые уравнения поправку в видеизотонического коэффициента i, учитывающего процесс распада на ионы молекул растворённого веществ Изотонический коэффициент (или фактор Вант-Гоффа) — это параметр, не имеющий размерности, который характеризует поведение какого – либо вещества в растворе. То есть, изотонический коэффициент показывает, разницу содержания частиц в растворе электролита по сравнению с раствором неэлектролита такой же концентрации. Он тесно связан связан с процессом диссоциации, точнее, со степенью диссоциации и выражается следующим выражением: i = 1+α(n—1), где:n – количество ионов, на которые диссоциирует вещество. α – степень диссоциации. Осмос – диффузия молекул растворителя через полупроницаемую мембрану из растворителя в раствор или из раствора с меньшей концентрацией в раствор с большей концентрацией. Осмотическое давление – величина измеряемая минимальным гидравлическим давлением, которое нужно приложить к раствору, чтобы осмос прекратился. Осмотическое давление раствора равно тому давлению, которое производило бы растворённое вещество, если бы оно при той же температуре находилось в газообразном состоянии и занимало объём, численно равный объёму раствора (закон Вант-Гоффа). Осмотическое давление и концентрацию раствора позволяет связать уравнение Вант — Гоффа, которое напоминает уравнение идеального газа Клапейрона – Менделеева: Pосм = CМ·R·T, где:CМ — молярная концентрация раствора, моль/л, R — универсальная газовая постоянная (8,314 Дж/моль·К); T — абсолютная температура раствора. Для растворов электролитов осмотическое давление определяется уравнением, в которое входит изотонический коэффициент: Pосм = i·CM·R·T, где: i — изотонический коэффициент раствора (для растворов электролитов i > 1, а для растворов неэлектролитов i = 1). Изотонический раствор Если полупроницаемой перегородкой разделены два раствора, имеющие одинаковое осмотическое давление, то перемещение растворителя через перегородку отсутствует. Гипотонический и гипертонический растворы Раствор, с меньшим осмотическим давлением, по сравнению с более концентрированным раствором, называют гипотоническим, а раствор с большей концентрацией – гипертоническим. 10)Сильные и слабые электролиты. Константа ионизации слабого электролита. Закон разведения Оствальда. Теория сильных электролитов Дебая-Хюккеля. Электролиты – это вещества, растворы и расплавы которых проводят электрический ток. Объяснение существования подвижных ионов в растворах электролитов дает теория электролитической диссоциации, сформулированная шведским ученым С. Аррениусом в первом варианте в 1883 г. после проведения количественного экспериментального исследования электропроводимости растворов. Электролитическая диссоциация - полный или частичный распад молекул растворенного вещества на катионы и анионы. Электролитической диссоциацией называют также распад на катионы и анионы ионных кристаллов при растворении или расплавлении. Электролиты: Сильные– практически полностью диссоциируют на ионы (>0,7). Например, HCl, HBr, HI, H2SO4, HNO3, растворимые соли, щелочи и др. Слабые– не полностью ионизируются (<0,1). Например, HCN, HF, CH3COOH, H2S, NH4OH, H2O и др. Законы Рауля и Вант-Гоффа не выполняются для растворов, которые проводят электрический ток – растворов электролитов.Повышение температуры кипения, понижение температуры замерзания, осмотическое давление для них всегда больше, чем у растворов неэлектролитов такой же концентрации. Эти аномалии впервые получили научное объяснение в разработанной Сванте Аррениусом теории электролитической диссоциации. Постулаты теории электролитической диссоциации Аррениуса: 1. Электролиты в растворах распадаются на катионы и анионы – диссоциируют. Распад молекул происходит не полностью и характеризуется степенью диссоциации α. Степенью диссоциации электролита называется отношение числа молекул, распавшихся на ионы, к общему числу его молекул, введённых в раствор, она характеризует силу электролита: α=n/N, где: n - число молекул, распавшихся на ионы; N - общее число растворённых частиц. 2. Диссоциация является обратимым равновесным процессом, к которому применим закон действующих масс и понятие константы диссоциации. Константа диссоциации – это константа равновесия, выраженная через концентрации образующихся ионов и непродиссоциировавших молекул. Константа диссоциации не зависит от концентрации, а лишь от природы вещества и температуры. 3. Силы взаимодействия ионов с молекулами растворителя и друг с другом малы (т.е. растворы являются идеальными). Закон разведения Оствальда:  Недостатки теории Аррениуса:Не объясняла причин электролитической диссоциации. Не учитывала взаимодействие с растворителем. Закономерности, полученные для слабых электролитов, не могут применяться к сильным электролитам без соответствующих поправок. Качественная теория сильных электролитов была разработана П. Дебаем и Г. Хюккелем (1923 г). Теория сильных электролитов Дебая-Хюккеля: Для сильных электролитов, даже при малой концентрации раствора энергия электростатического взаимодействия между ионами достаточно велика. Противоположно заряженные ионы отталкиваются, одноименно заряженные ионы притягиваются. В результате вокруг каждого иона находятся преимущественно ионы с противоположным зарядом – ионная атмосфера. Радиус ионной атмосферы сравнительно велик, поэтому ионные атмосферы соседних ионов пересекаются; кроме того, каждый ион окружен дипольными молекулами растворителя – сольватной оболочкой. Т.е., в растворе сильного электролита возникает подобие пространственной структуры, что ограничивает свободу перемещения ионов и изменяет свойства раствора так, будто бы уменьшилась степень диссоциации. Поэтому, определяя степень диссоциации раствора сильного электролита, получают кажущуюся степень диссоциации, т.е. величину α с поправкой на межионное взаимодействие. Чем выше концентрация раствора, тем сильнее взаимодействие ионов, тем меньше кажущаяся степень диссоциации сильного электролита. Количественные расчеты характеристик растворов сильных электролитов осуществляют с помощью понятия активности электролита. Активность (α)- это эффективная (действительная) концентрация электролита, учитывающая электростатическое взаимодействие ионов в растворе. α = fC, где: α – активность иона; C – его концентрация; f – коэффициент активности. Значение f < 1 указывает на связывающее взаимодействие ионов; если f близок к единице, это говорит о слабом межионном взаимодействии. В очень разбавленных растворах действие межионных сил почти не проявляется. Физический смысл коэффициента активности f: показывает меру отклонения свойств реального раствора от идеального. 11)Ионная сила раствора. Активность и коэффициент активности ионов. Электролиты в организме. Осмоляльность и осмолярность биологических жидкостей и перфузионных растворов. Гипо-, гипер- и изотоничные растворы. Изотонический коэффициент. Понятие об электролитном гомеостазе (изоосомии). Роль осмоса в биологических системах. Плазмолиз и цитолиз. Ионная сила раствора – величина, измеряемая полусуммой произведений концентраций всех находящихся в растворе ионов на квадрат их заряда. I=0.5⋅∑СiZi^2, где:Ci– молярная концентрация иона в растворе, моль/л; Zi– заряд иона. Активность иона – это его эффективная концентрация. Активность связана с молярной концентрацией следующим образом: a=f*СM, где f – коэффициент активности – величина, показывающая во сколько раз активность ионов отличается от их истинной аналитической концентрации (Сi) в растворе сильного электролита. Величина f зависит от природы, температуры и концентрации электролитов в растворе. Для разбавленных растворов электролитов можно принять, что f=1,т.к. межионные взаимодействия практически отсутствуют и величина f в этом случае зависит от концентрации и заряда ионов, но практически не зависит от их природы. С увеличением концентрации величина f уменьшается, т.к. уплотняется ионная атмосфера. Для высоконцентрированных растворов f)>>1, т.к. гидратная оболочка ионов практически отсутствует и их подвижность возрастает. Рассчитать величину f можно по уравнению Дебая-Хюккеля: lg f =–A⋅Zi^2⋅квадратный корень из I, где:А – коэффициент, зависящий от температуры; I-ионная сила раствора;Zi – заряд иона. Электролиты в организме: Na и Cl участвуют в поддержании кислотно-щелочного баланса, осмотического равновесия в организме. Са играет большую роль в построении костной ткани и зубов, в регулировании кислотности крови и ее свертывании, в возбудимости мышечной и нервной ткани. К находится преимущественно в жидкостях тела и мягких тканях, где является необходимым элементом для поддержания осмотического давления, регуляции рН крови. Mg является кофактором многих ферментативных реакций, необходим на всех этапах синтеза белка. В живых организмах Fe является важным микроэлементом, катализирующим процессы обмена кислородом. Сo входит в состав витамина В12, задействован при кроветворении, функциях нервной системы и печени, ферментативных реакциях. Zn необходим для метаболизма витамина E, участвует в синтезе разных анаболических гормонов в организме, включая инсулин, тестостерон и гормон роста. Mn оказывает влияние на рост, образование крови и функции половых желёз. Концентрация ионов в биологических жидкостях поддерживается примерно постоянной (ионный гомеостаз), поэтому приём и выделение солей тесно связаны с объёмом воды. В норме в организме имеется постоянный баланс между поступлением и потерей жидкости. При нарушении водного обмена могут возникнуть дегидратация и гипергидратация. Осмолярность (осмоляльность) – активная концентрация частиц, не проникающих через идеальную полупроницаемую мембрану. Для разбавленных растворов их численные значения совпадают. Это эмпирические величины, использование которых позволяет учесть разные по характеру отклонения от закона Рауля, возникающие в случае неидеальных растворов. Осмоляльность крови в значительной степени зависит от концентрации ионов натрия и хлора, в меньшей степени глюкозы и мочевины. В норме осмоляльность сыворотки крови 275— 296 мосмоль/кг Н20, осмоляльность мочи обусловлена мочевиной, ионами натрия, калия, аммония. Осмоляльность мочи колеблется значительно: от 50 до 1400 мосмоль/кг Н20. При суточном диурезе около 1,5 л осмоляльность мочи здорового человека составляет 600—800 мосмоль/кг Н20. Изотонический раствор. Если полупроницаемой перегородкой разделены два раствора, имеющие одинаковое осмотическое давление, то перемещение растворителя через перегородку отсутствует. Гипотонический и гипертонический растворы. Раствор, с меньшим осмотическим давлением, по сравнению с более концентрированным раствором, называют гипотоническим, а раствор с большей концентрацией – гипертоническим. Изотонический коэффициент (или фактор Вант-Гоффа) — это параметр, не имеющий размерности, который характеризует поведение какого – либо вещества в растворе. То есть, изотонический коэффициент показывает, разницу содержания частиц в растворе электролита по сравнению с раствором неэлектролита такой же концентрации. Он тесно связан связан с процессом диссоциации, точнее, со степенью диссоциации и выражается следующим выражением: i = 1+α(n—1), где: n – количество ионов, на которые диссоциирует вещество. α – степень диссоциации. Изоосмия (электролитный гомеостаз) - относительное постоянство осмотического давления в жидких средах и тканях организма, обусловленное поддержанием на данном уровне концентраций содержащихся в них веществ: электролитов, белков и т. д. Это одна из важнейших физиологических констант организма, обеспечиваемых механизмами саморегуляции (гомеостаза). Явление осмоса играет важную роль во многих химических и биологических системах. Благодаря осмосу регулируется поступление воды в клетки и межклеточные структуры. Упругость клеток (тургор), обеспечивающая эластичность тканей и сохранение определенной формы органов, обусловлена осмотическим давлением. Животные и растительные клетки имеют оболочки или поверхностный слой протоплазмы, обладающие свойствами полупроницаемых мембран. При помещении этих клеток в растворы с различной концентрацией наблюдается осмос. Плазмолиз - отставание протопласта от оболочки при погружении клетки в гипертонический раствор. Характерен для растительных клеток. Животные клетки сжимаются. Цитолиз - разрушение животных и растительных клеток, выражающееся в полном или частичном их растворении. 12)Предмет и основные понятия химической кинетики. Химическая кинетика как основа для изучения скоростей и механизмов биохимических процессов. Скорость реакции, средняя скорость реакции в интервале, истинная скорость. Зависимость скорости реакции от концентрации. Химическая кинетика изучает скорости химических реакций, их зависимость от различных факторов и механизмы реакций. Последовательность и характер стадий химических реакций называют механизмом реакции. Изучение кинетики и механизма химических реакции имеет большое теоретическое и практическое значение как в химии, так в биологии. Как известно, химические реакции могут протекать с самыми различными скоростями. Некоторые реакции, сопровождающиеся взрывом, заканчиваются в тысячные доли секунды, другие же совершаются в течение минут, часов или даже многих лет, например геохимические процессы в земной коре. Кинетика имеет большое практическое значение для проведения различных технологических процессов. Скорость этих процессов может быть изменена в желаемом направлении в зависимости от создаваемых условий. Особенности течения биохимических процессов зависят от катализаторов; эффективность действия лекарственных веществ может быть связана со скоростью химических реакций, возникающих при этом в организме, и т. п. Основной постулат химической кинетики – закон действующих масс. Скоростью гомогенной химической реакции называется изменение количества реагентов (или продуктов реакции) в единицу времени в единице объёма. V=±ΔС/Δt, где:ΔС - изменение концентрации одного из реагирующих веществ; Δt - промежуток времени. Средняя скорость в интервале – изменение концентрации реагирующих веществ за некоторый промежуток времени. Истинная скорость – изменение концентрации реагирующих веществ в конкретный момент времени и представляет собой предел средней скорости при Δt→0 Зависимость скорости от концентрации реагирующих веществ определяется основным законом действующих масс: скорость химической реакции прямо пропорциональна произведению концентраций взаимодействующих веществ, взятых в степенях их стехиометрических коэффициентов. Математическая зависимость скорости реакции от концентрации реагентов называется кинетическим уравнением. V = k[A]^2⋅[B]^2, где: k - константа скорости химической реакции. 13)Классификации реакций, применяющиеся в кинетике: реакции гомогенные, гетерогенные и микрогетерогенные; реакции простые и сложные (параллельные, последовательные, сопряжённые, цепные). Период полувыведения. Химическая кинетика изучает скорости химических реакций, их зависимость от различных факторов и механизмы реакций. Последовательность и характер стадий химических реакций называют механизмом реакции. По механизму различают: Простыми называют реакции, протекающие в одну стадию. Сложными называют реакции, протекающие в несколько стадий. Выделяют следующие виды сложных реакций: Параллельные - характерно протекание нескольких процессов с участием одних и тех же исходных веществ. Эти процессы завершаются образованием разных продуктов реакции. Скорость параллельных реакций определяется наиболее быстрой стадией; Последовательные - образование конечного продукта реакции из исходных веществ происходит не непосредственно, а через ряд промежуточных продуктов. Скорость последовательной реакции определяется наиболее медленной стадией, которая называется лимитирующей; Сопряженные Цепные. По фазовому состоянию реагентов реакции бывают гомогенные (однородные) и гетерогенные (неоднородные). В гомогенных реакциях все взаимодействующие вещества находятся в одной фазе (газовой, жидкой или твердой). Зоной реакции при проведении гомогенных реакций служит весь реакционный объем. В гетерогенных процессах реагенты, принимающие участие в реакции, находятся в разных фазах. В реакционном объеме одновременно находятся две или более фазы, а химическая реакция протекает на границе раздела фаз или в объеме одной из фаз. Выделяют такжемикрогетерогенные реакции - реагенты находятся в коллоидном или высокомолекулярном состоянии. Гетерогенные двухфазные реакции в зависимости от агрегатного состояния исходных веществ бывают следующих типов: -В системе «газ – твердое тело»; -Между двумя несмешивающимися жидкостями; -В системе «газ – жидкость»; -В системе «жидкость – твердая фаза». Период полувыведения вещества (биологический период полувыведения) — время, нужное веществу (например метаболиту, биологически активному веществу, радионуклиду, и т. д.) для потери половины его фармакологического, физиологического или радиоактивного действия. Как правило, это относится к очищению организма через функцию почек и печени в дополнение к функции экскреции и удалению вещества из организма. 14)Зависимость скорость реакции от температуры. Температурный коэффициент скорости реакции и его особенности для биохимических процессов. Понятие о теории активных соударений. Энергия активации; уравнение Аррениуса. Понятие о теории переходного состояния. Зависимость скорости реакций от температуры определяется правилом Вант-Гоффа: При увеличении температуры на каждые 10°С скорость большинства реакций увеличивается приблизительно в 2—4 раза. Уравнение Вант-Гоффа: V(T2) = V(T1) ⋅ y^(T2-T1)/10 где у — температурный коэффициент константы скорости реакции, или коэффициент Вант-Гоффа. Многие каталитические реакции не подчиняются этому правилу, для большинства из них у < 2. Почти все биохимические реакции осуществимы только в присутствии ферментов, следовательно, почти все они не подчиняются данному правилу. Кроме того, при увеличении температуры свыше определенного значения (45—50°С) биохимические реакции резко замедляются, а затем останавливаются, что связано с инактивацией ферментов при высоких температурах. Более точную зависимость скорости химической реакции от температуры устанавливает уравнение Аррениуса: k = Ae^(-Ea/RT), где: k - константа скорости реакции; A - константа Аррениуса; Еа - энергия активации (Дж/моль); R - универсальная газовая постоянная (8,31); T - абсолютная температура в кельвинах; e - основание натурального логарифма. Энергия активации реакции Еа — энергетический барьер, который должны преодолеть исходные вещества по пути превращения в продукты реакции, зависит от природы реагирующих веществ и служит характеристикой каждой реакции. Согласно теории активных соударений химическая реакция происходит при каждом столкновении реагирующих частиц, соответствующим образом ориентированных относительно друг друга и обладающих энергией, равной (или больше) энергии активации. В теории активных соударений используются основные положения молекулярно-кинетической теории и предположения о существовании энергетического барьера, который должен быть преодолен в ходе химической реакции. В простейшем случае теория активных столкновений предполагает, что для осуществления реакции молекулы должны сблизиться на расстояние, равное полусумме их эффективных диаметров, учитывающих взаимодействие, при этом молекулы рассматриваются как жесткие сферы. Теория переходного состояния постулирует, что реагирующие молекулы сначала образуют переходный «активированный комплекс», находящийся в равновесии с исходными веществами. Затем этот комплекс превращается в конечные продукты, причем скорость реакции определяется именно скоростью разложения переходного комплекса X: А+В→[X]→C+D где X — переходный комплекс, часто обозначаемый звездочкой: [Х]=[АВ]*. Доказательство в теории переходного состояния того факта, что скорость реакции зависит не только от энергии активации, но и от энтропии активации, позволяет объяснить различие в скоростях реакций с близкими величинами энергии активации. Скорость будет выше у той реакции, энтропия активации которой больше. 15)Катализ. Гомогенный и гетерогенный катализ. Энергетический профиль каталитической реакции. Особенности каталитической реакции. Особенности каталитической активности ферментов. Катализом называют явление изменения скорости химической реакции под воздействием катализаторов. Реакции, протекающие с участием катализаторов, называют каталитическими. Если от добавления катализатора к реагирующей смеси скорость реакции увеличивается, катализ называют положительным, если же реакция замедляется, то катализ называют отрицательными, а катализатор ингибитором. Принципы катализа (особенности каталитической реакции): -Катализатор принимает участие в химической реакции, образуя промежуточные продукты, но в конце реакции выделяется в химически неизменном виде. Физическое состояние катализатора, входящего в активный комплекс, может существенно изменяться, например, уменьшатся размеры зерен твердого катализатора, изменится структура поверхностных слоев; -Катализатор не смещает положение равновесия, а лишь увеличивает скорость прямой и обратной реакции в равной степени; -Действие катализатора является специфичным (селективным); -Катализатор увеличивает скорость реакции за счет уменьшения Еа, ведет реакцию по пути с меньшим энергетическим барьером - энергетический профиль каталитической реакции. Гомогенный – это такой катализ, когда катализатор и все реагирующие вещества находятся в одной фазе. Гомогенный катализ в растворах наиболее часто вызывается действием водородных и гидроксильных ионов. Каталитическое действие кислот было открыто в 1811 г. К. Кирхгофом. Инверсия сахара, омыление сложных эфиров, гидролитическое разложение амидов, ацеталей и много других реакций в растворах ускоряются действием водородных ионов, причем с повышением их концентрации примерно пропорционально увеличивается и скорость.Главным положением гомогенного катализа является представление о том, что в ходе реакции образуются неустойчивые промежуточные соединения катализатора с реагирующими веществами, которые затем распадаются с регенерацией катализатора. К гомогенному катализу относятся многие реакции кислотно-основного взаимодействия, реакции комплексообразования, многочисленные реакции гидрирования, сульфидирования, реакции, катализированные ферментами. К гетерогенным относятся каталитические процессы, протекающие на границе раздела фаз Т-Г, Т-Ж. При гетерогенном катализе реакция протекает на поверхности катализатора. Поэтому площадь поверхностного слоя катализатора и его строение определяют активность катализатора. Гетерогенный катализ находит большее применение в промышленности, чем гомогенный. В качестве гетерогенных катализаторов используют переходные металлы, металлы первой группы, фосфорную кислоту. Энергетический профиль каталитической реакции:  Ферментативными реакциями называются такие химические процессы в биологических системах, скорость которых регулируется веществами биологического происхождения. Это белковые молекулы, называемые ферментами или энзимами. Ферментативный катализ играет огромную роль в жизнедеятельности организма. Широкое применение получили ферментные препараты при нарушениях функции желудочно-кишечного тракта, связанных с недостаточной выработкой пищеварительных ферментов (пепсин, панкреатин). При ожогах, гнойных ранах, гнойно-воспалительных заболеваниях легких, когда необходимо разрушить накопившиеся в большом количестве белковые образования, применяются протолитические ферменты, приводящие к быстрому гидролизу белков и способствующие рассасыванию гнойных скоплений. Для лечения инфекционных заболеваний используются препараты лизоцина, которые разрушают оболочку некоторых болезнетворных бактерий. Очень важные ферменты, которые рассасывают тромбы (сгустки крови внутри кровеносных сосудов) – плазмин, трипсин, химотрипсин, на их основе с разными добавками созданы различные лекарственные препараты – стрептокиназа, стрептаза, и т.п., широко применяемые в медицине. Выделения ферментов в особый класс катализаторов обусловлен особыми свойствами этих веществ: Высокая специфичность; Эффективность действия; Биологические катализаторы образуются и разрушаются в процессе жизнедеятельности организма. По своей каталитической активности биологические катализаторы в тысячи раз превышают неорганические.Специфичность действия связана с особенностями структуры фермента и субстрата. Одни части каталитической системы выполняют функции, главным образом связанные с пространственной организацией системы, другие в этой организационной системе осуществляют собственно катализ. Т.е., как и при неферментативном катализе, в каталитической реакции участвует не вся белковая молекула в целом, а лишь определенные ее участки – активные центры фермента. |