биохимия лекции. Энзимология как учение о ферментах. Простые и сложные ферменты
 Скачать 3.9 Mb. Скачать 3.9 Mb.
|

|
Раздел 4.4 Единицы активности ферментов, их применение. Международная комиссия по ферментам предложила за единицу активности любого фермента принимать такое количество фермента, которое при заданных условиях катализирует превращение одного микромоля (10–6 моль) субстрата в единицу времени (1 мин, 1 час) или одного микроэквивалента затронутой группы в тех случаях, когда атакуется более одной группы в каждой молекуле субстрата (белки, полисахариды и другие). Должна быть указана температура, при которой проводится реакция. Результаты измерений активности ферментов могут быть выражены в единицах общей, удельной и молекулярной активности. Общая активность фермента За единицу общей активности фермента принимают такое количество фермента, которое катализирует превращение 1 мкмоль субстрата в единицу времени в расчёте на количество материала, взятого для исследования. Так, активность аланинаминотрансферазы в печени крыс равна 1670 мкмоль пирувата в час на 1 г ткани; активность холинэстеразы в сыворотке крови человека составляет 250 мкмоль уксусной кислоты в час на 1 мл сыворотки при 37°C. Особого внимания исследователя требуют высокие значения активности фермента как в норме, так и в патологии. Рекомендуется работать с небольшими показателями активности фермента. Для этого источник фермента берут в меньшем количестве (сыворотку разводят в несколько раз физиологическим раствором, а для ткани готовят меньший процентный гомогенат). По отношению к ферменту в таком случае создаются условия насыщения субстратом, что способствует проявлению его истинной активности. Общая активность фермента рассчитывается с помощью формулы:  где а – активность фермента (общая), ΔС – разность концентраций субстрата до и после инкубации; В – количество материала, взятого на анализ, t - время инкубации; n - разведение. Следует иметь в виду, что показатели активности ферментов сыворотки крови и мочи, исследуемых в диагностических целях, выражают в единицах общей активности. Удельная активность фермента Поскольку ферменты являются белками, важно знать не только общую активность фермента в исследуемом материале, но и ферментативную активность белка, находящегося в данной пробе. За единицу удельной активности принимают такое количество фермента, которое катализирует превращение 1 мкмоль субстрата в единицу времени в расчёте на 1 мг белка пробы. Для вычисления удельной активности фермента необходимо общую активность разделить на содержание белка в пробе:  Например, содержание белка в ткани печени составляет 160 мг/г. Разделив общую активность аланинаминотрансферазы (см. выше) на это значение, получаем 10,4 мкмоль пирувата/мг белка × час. Чем хуже очищен фермент, тем больше в пробе находится посторонних балластных белков, тем ниже удельная активность. В ходе очистки количество таких белков уменьшается, и соответственно удельная активность фермента повышается. Предположим, в исходном биологическом материале, являющемся источником фермента (измельчённая печень, кашица из растительной ткани), удельная активность была равна 0,5 мкмоль/ (мг белка× мин). После дробного осаждения сульфатом аммония и гель-фильтрации через сефадекс она повысилась до 25 мкмоль/ (мг белка× мин), т.е. увеличилась в 50 раз. К оценке эффективности очистки ферментных препаратов прибегают при производстве лекарственных средств энзиматической природы. Удельную активность определяют в том случае, когда нужно сопоставить активность разных препаратов одного и того же фермента. Если требуется сравнить активность разных ферментов, рассчитывают молекулярную активность. Молекулярная активность фермента Молекулярная активность (или число оборотов фермента) – это количество моль субстрата, подвергающееся превращению под действием 1 моль фермента в единицу времени (обычно в 1 минуту). Разным ферментам присуща неодинаковая молекулярная активность. Уменьшение числа оборотов ферментов происходит под действием неконкурентных ингибиторов. Изменяя конформацию каталитического центра фермента, эти вещества понижают сродство фермента к субстрату, что приводит к уменьшению числа молекул субстрата, реагирующих с одной молекулой фермента в единицу времени. Раздел 4.5 Энзимодиагностика. Некоторые ферменты, проферменты и их субстраты в норме постоянно циркулируют в крови человека и выполняют физиологические функции. Примерами таких ферментов плазмы являются липопротеинлипаза, псевдохолинэстераза, а также проферменты компонентов систем свертывания крови и растворения кровяного сгустка. Эти ферменты называются секреторными, большинство из них синтезируется в печени и секретируются в кровь. Концентрация таких энзимов в крови либо такая же, как в тканях, либо более высокая. Кроме секреторных ферментов, в плазме крови присутствуют ферменты, которые не выполняют в крови никаких известных физиологических функций. Их субстраты в плазме обычно не обнаруживаются, и в норме их концентрация в крови человека почти в миллион раз ниже, чем в тканях. Такие ферменты называются индикаторными. Появление их в плазме крови в повышенных концентрациях указывает на повышенную скорость разрушения тканей. Таким образом, измерение в крови уровня индикаторных ферментов плазмы дает врачу ценную диагностическую и прогностическую информацию. Индикаторные ферменты обычно обнаруживаются в плазме в малых количествах, и появляются в ней вследствие постоянно протекающих процессов разрушения клеток организма. Появление этих ферментов в плазме в повышенных концентрациях указывает на повышенную скорость разрушения тканей. Поступлением в плазму значительных количеств мышечных ферментов сопровождается и выполнение тяжелой физической работы. Для клеток разных органов характерен свой набор ферментов, поэтому повышение в крови активности того или иного фермента может указывать на заболевание соответствующего органа. В клинической практике используется количественное определение различных индикаторных ферментов плазмы. В клетках поджелудочной железы вырабатываются ферменты трипсин (трипсиноген), липаза и амилаза; при остром воспалении поджелудочной железы (острый панкреатит) активность этих ферментов в крови возрастает; повышенная активность амилазы обнаруживается и в моче. Довольно часто в целях диагностики проводят анализ изоферментного спектра некоторых ферментов, в частности ЛДГ. В клетках миокарда преобладает изофермент ЛДГ1. При заболеваниях, связанных с повреждением сердечной мышцы, концентрация и активность этого изофермента в плазме крови значительно возрастает. При некоторых заболеваниях печени (в том числе инфекционной природы) в крови возрастает содержание ЛДГ4 и ЛДГ5 - изоферментов, характерных для клеток печени. В настоящее время для получения этой ценной диагностической и прогностической информации в большинстве случаев используются автоанализаторы. В таблице 4.1 приведен перечень ферментов, активность которых чаще всего исследуют в диагностической энзимологии. Таблица 4.1. Основные ферменты сыворотки, используемые в клинической диагностике. Аспартатаминотрансфераза Инфаркт миокарда Аланинаминотрансфераза Вирусный гепатит Амилаза Острый панкреатит Церулоплазмин Гепатолентикулярная дегенерация (болезнь Вильсона) Креатинфосфокиназа Заболевание мышц и инфаркт миокарда γ-Глутамилтранспептидаза Различные заболевания печени Лактатдегидрогеназа (изозимы) Инфаркт миокарда Липаза Острый панкреатит Кислая фосфатаза Рак предстательной железы Щелочная фосфатаза (изозимы) Различные заболевания костей, закупорка протоков печени Высокая специфичность ферментов позволяет использовать их для обнаружения и количественного определения нормальных и патологических компонентов крови и мочи. Например, с помощью фермента уреазы, действующего только на мочевину, можно проводить определение этого вещества в биологических жидкостях. Фермент глюкозооксидаза применяется для определения глюкозы в крови и моче у больных сахарным диабетом. Раздел 5.1 Энзимопатии: причины, проявления, методы диагностики. В 1908 году английский врач Арчибальд Гаррод высказал предположение, что причиной ряда заболеваний может являться отсутствие какого-либо из ключевых ферментов, участвующих в обмене веществ. Он ввёл понятие "inborn errors of metabolism" (врождённый дефект обмена веществ). В дальнейшем эта теория была подтверждена новыми данными, полученными в области молекулярной биологии и патологической биохимии. Информация о последовательности аминокислот в полипептидной цепи белка записана в соответствующем участке молекулы ДНК в виде последовательности тринуклеотидных фрагментов - триплетов или кодонов. Каждый триплет кодирует определённую аминокислоту. Такое соответствие называется генетическим кодом. Причём некоторые аминокислоты могут быть закодированы при помощи нескольких кодонов. Существуют также специальные кодоны, являющиеся сигналами для начала синтеза полипептидной цепи и его прекращения. К настоящему времени генетический код полностью расшифрован (таблица 5.1). Он является универсальным для всех видов живых организмов. Таблица 5.1 Триплетный код нуклеотидов мРНК для аминокислот  Примечания. * - этот кодон является также сигналом для начала синтеза полипептидной цепи (инициирующий кодон); ** - эти кодоны не соответствуют ни одной из аминокислот и служат сигналом для прекращения синтеза полипептидной цепи (терминирующие кодоны). Реализация информации, заложенной в молекуле ДНК, включает несколько этапов. Сначала в клеточном ядре в процессе транскрипции синтезируется матричная РНК (мРНК), поступающая в цитоплазму. В свою очередь, мРНК служит матрицей для трансляции - синтеза полипептидных цепей на рибосомах. Таким образом, природа молекулярных болезней определяется нарушением структуры и функции нуклеиновых кислот и контролируемых ими белков. Мутации Поскольку информация о структуре всех белков клетки содержится в последовательности нуклеотидов ДНК, а каждая аминокислота определяется триплетом нуклеотидов, изменение первичной структуры ДНК может в конечном счёте оказать глубокое влияние на синтезируемый белок. Подобные изменения происходят за счёт ошибок репликации ДНК, когда одно азотистое основание заменяется другим, либо в результате действия радиации или при химической модификации. Все возникшие таким образом наследуемые дефекты называются мутациями. Они могут приводить к неправильному считыванию кода и делеции (выпадению) ключевой аминокислоты, замене одной аминокислоты другой, преждевременной остановке белкового синтеза или добавлению аминокислотных последовательностей. Учитывая зависимость пространственной упаковки белка от линейной последовательности в нём аминокислот, можно полагать, что подобные дефекты способны изменить структуру белка, а значит, и его функцию. Тем не менее, многие мутации обнаруживаются только в лабораторных условиях и не оказывают вредного воздействия на функции белка. Таким образом, ключевым моментом является локализация изменений в первичной структуре. Если положение замененной аминокислоты окажется критическим для формирования третичной структуры и образования каталитического центра фермента, то мутация является серьёзной и может проявиться как заболевание. Последствия для обмена веществ Последствия недостаточности одного фермента в цепи реакций обмена веществ могут проявляться по-разному. Предположим, что превращение соединения A в соединение B катализирует фермент Е и что соединение C встречается на альтернативном пути превращений (рисунок 5.1):  Рисунок 5.1. Схема альтернативных путей биохимических превращений. Последствиями недостаточности фермента могут быть следующие явления: недостаточность продукта ферментативной реакции (B). В качестве примера можно указать на снижение содержания глюкозы в крови при некоторых формах гликогенозов; накопление вещества (A), превращение которого катализирует фермент (например, гомогентизиновая кислота при алкаптонурии). При многих лизосомных болезнях накопления, вещества, в норме подвергающиеся гидролизу в лизосомах, накапливаются в них в связи с недостаточностью одного из ферментов; отклонение на альтернативный путь с образованием некоторых биологически активных соединений (C). К этой группе явлений относится экскреция с мочой фенилпировиноградной и фенилмолочной кислот, образующихся в организме больных фенилкетонурией в результате активации вспомогательных путей распада фенилаланина. Если метаболическое превращение в целом регулируется по принципу обратной связи конечным продуктом, то эффекты двух последних типов аномалий будут более значительными. Так, например, при порфириях (врождённых нарушениях синтеза гема) устраняется подавляющего эффекта гема на начальные реакции синтеза, что приводит к образованию избыточных количеств промежуточных продуктов метаболического пути, которые обладают токсическим действием на клетки кожи и нервной системы. Факторы внешней среды могут усиливать или даже полностью определять клинические проявления некоторых врождённых нарушений обмена веществ. Например, у многих пациентов с недостаточностью глюкозо-6-фосфатдегидрогеназы заболевание начинается только после приёма таких лекарственных средств, как примахин. В отсутствие контактов с лекарственными средствами такие люди производят впечатление здоровых. Лабораторная диагностика врождённых нарушений обмена веществ О недостаточности фермента обычно судят косвенно по повышению концентрации исходного вещества, которое в норме подвергается превращениям под действием данного фермента (например, фенилаланин при фенилкетонурии). Прямое определение активности таких ферментов проводят только в специализированных центрах, но по возможности диагноз следует подтверждать этим методом. Пренатальная (дородовая) диагностика некоторых врождённых нарушений метаболизма возможна путём иследования клеток амниотической жидкости, полученных на ранних стадиях беременности и культивируемых in vitro. Лечение при врождённых нарушениях метаболизма Некоторые врождённые нарушения метаболизма поддаются лечению путём доставки в организм недостающего метаболита или путём ограничения поступления в желудочно-кишечный тракт предшественников нарушенных процессов обмена веществ. Иногда могут быть удалены накапливающиеся продукты (например, железо при гемохроматозе). Раздел 5.2 Наиболее распространённые энзимопатии. Врождённые нарушения обмена фенилаланина и тирозина На рисунке 5.2 представлена схема основных химических превращений фенилаланина и тирозина, а также известных в настоящее время нарушений активности ферментов, катализирующих эти реакции. Из схемы видно, что тирозин, который в норме образуется в организме из фенилаланина, является предшественником целого ряда биологически активных соединений. 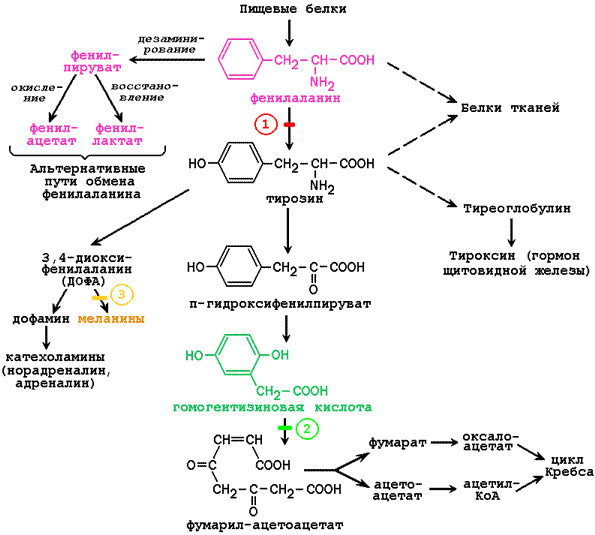 Рисунок 5.2. Обмен фенилаланина и тирозина и возможные нарушения. Цифрами показаны блокированные реакции при следующих заболеваниях: 1 - фенилкетонурия; 2 - алкаптонурия; 3 - альбинизм. Фенилкетонурия. Это заболевание вызывается дефицитом печёночного фермента фенилаланингидроксилазы, или реже, нарушением биосинтеза его кофактора тетрагидробиоптерина. Поскольку фенилаланингидроксилаза катализирует превращение фенилаланина в тирозин, при фенилкетонурии в крови накапливается фенилаланин, который наряду с продуктами его альтернативных превращений (фенилпируват, фениллактат, фенилацетат) экскретируется с мочой. Присутствие в моче пациентов фенилпирувата (соединения, содержащего кетогруппу) нашло отражение в названии заболевания. Избыток фенилаланина вызывает замедление транспорта тирозина и других аминокислот через клеточные мембраны. Следствием этого может быть нарушение обмена аминокислот в клетках головного мозга с последующим расстройством биосинтеза белка и нарушением синтеза нейромедиаторов. Если своевременно не выявить дефект фермента и не начать лечение, в течение первого года жизни у детей развивается умственная отсталость. В связи с этим в ряде стран широко практикуются скрининговые исследования новорождённых с целью раннего выявления случаев фенилкетонурии (определение концентрации фенилаланина в крови, взятой из прокола кожи на пятке). Лечение заболевания заключается в том, чтобы ограничить поступление фенилаланина с пищей. С этой целью используются искусственные питательные смеси. По современным рекомендациям, длительность лечения фенилкетонурии составляет от 5 до 10 лет. Алкаптонурия. Это заболевание обусловлено врождённой недостаточностью оксидазы гомогентизиновой кислоты. Гомогентизиновая кислота накапливается в крови, тканях и моче. Окисление и полимеризация этого вещества приводит к образованию пигмента алкаптона. Отложение алкаптона в хрящах, которые затем темнеют, называется охронозом. Превращению гомогентизиновой кислоты в алкаптон способствует щелочная среда; при алкаптонурии наиболее явным симптомом является экскреция либо чёрной мочи, либо мочи, которая темнеет по мере защелачивания при хранении. Алкаптонурия в большинстве случаев не требует специального лечения, но в среднем возрасте и позже обычно развивается артрит. Альбинизм. Недостаточность фермента тирозиназы в меланоцитах (пигментных клетках кожи и радужной оболочки глаз) вызывает одну из форм альбинизма и наследуется как рецессивный признак. У пациентов отсутствует пигментация кожи, волос и радужной оболочки (глаза кажутся розовыми). Отсутствие пигмента в коже сопровождается повышенной чувствительностью к солнечным лучам. Следует отметить, что биосинтез адреналина у альбиносов не нарушается, так как тирозиназа, участвующая в образовании катехоламинов, представляет собой другой фермент, контролируемый иным геном. Галактоземия Галактоземия - врождённое нарушение обмена веществ, обусловленное недостаточностью фермента галактозо-1-фосфатуридилтрансферазы. Для галактоземии характерна триада симптомов: увеличение размера печени, катаракта и умственная отсталость. В крови больных повышено содержание галактозы, этот моносахарид обнаруживается и в моче. Первые признаки заболевания у ребёнка (диарея, рвота, обезвоживание) выявляются очень рано, обычно через несколько дней после начала грудного вскармливания. Источником галактозы в организме является дисахарид лактоза, содержащаяся в молоке. После расщепления лактозы в микроворсинках слизистой оболочки тонкого кишечника галактоза под действием фермента галактокиназы превращается в печени в галактозо-1-фосфат (рисунок 5.3).  Рисунок 5.3. Обмен галактозы и основная причина галактоземии. В нормальных условиях галактозо-1-фосфат под влиянием галактозо-1-фосфатуридилтрансферазы переходит в УДФ-галактозу. В результате угнетения этой реакции в организме накапливается галактозо-1-фосфат - метаболит с очень коротким в нормальных условиях периодом существования. В связи с этим в норме он не вызывает нарушений. Однако при накоплении галактозо-1-фосфата проявляется его мощное токсическое действие. Природа токсического влияния галактозо-1-фосфата, вероятно, объясняется структурным сходством галактозы с глюкозой. Галактозо-1-фосфат, присоединяясь к активному центру ферментов, метаболизирующих глюкозо-1-фосфат, блокирует их, что приводит к нарушению обмена глюкозы. Так, в клетках печени накопление галактозо-1-фосфата вызывает ингибирование фосфоглюкомутазы и глюкозо-6-фосфатазы - ферментов, участвующих в превращении гликогена в глюкозу, в результате чего снижается уровень глюкозы в крови. В хрусталике глаза избыток галактозы переходит в шестиатомный спирт галактит. Галактит не подвергается дальнейшим превращениям и приводит к набуханию соединительной ткани и развитию катаракты. В клетках головного мозга нарушается синтез гликолипидов вследствие недостаточного образования их предшественника УДФ-галактозы. Если галактозу не исключить из диеты, возможны тяжёлые последствия, в том числе летальный исход. Гликогенозы Этот термин является общим для группы наследственных заболеваний, характеризующихся отложением в тканях аномально больших количеств полисахарида - гликогена, являющегося важным источником энергии и резервом углеводов в тканях. Врождённые нарушения содержания и структуры гликогена обусловлены дефицитом одного из ферментов, участвующих в расщеплении гликогена в печени или в скелетных мышцах (рисунок 5.4).  Рисунок 5.4. Расщепление гликогена в печени и скелетных мышцах и его нарушения. Примеры: Гликогеноз I типа (болезнь Гирке) – дефицит глюкозо-6-фосфатазы в печени. Характеризуется повышенным содержанием гликогена в печени; содержание глюкозы в крови снижено. После введения адреналина или глюкагона (гормонов, активирующих фермент гликогенфосфорилазу), уровень пирувата и лактата в крови существенно возрастает. Гликогеноз V типа (болезнь Мак-Ардля) – дефицит фосфорилазы в скелетных мышцах. У больных развивается пониженная выносливость к физическим нагрузкам. В скелетных мышцах содержится аномально высокое количество гликогена. Тем не менее, после выполнения физической работы или после введения адреналина содержание лактата в крови не увеличивается. Гликогеноз VI типа (болезнь Херса) – дефицит фосфорилазы в печени. Для этого заболевания характерно повышение содержания гликогена в печени, гипогликемия. После введения адреналина или глюкагона содержание лактата в крови не увеличивается (в отличие от гликогеноза I типа). |