Билеты по химии. История представления об атомах. Состав и строение атома. Ученье Демокрита
 Скачать 276.97 Kb. Скачать 276.97 Kb.
|

|
Основным законом химической кинетики является постулат, вытекающий из большого числа экспериментальных данных и выражающий зависимость скорости реакции от концентрации. Этот закон называют законом действующих масс. Он утверждает, что скорость химической реакции в каждый момент времени пропорциональна концентрациям реагирующих веществ, возведённым в некоторые степени. Если уравнение химической реакции имеет вид: a A + b B + d D → продукты, то формулу закона действующих масс можно представить в виде: 24.Влияние температуры на скорость химической реакции Температура является очень важным фактором, определяющим скорость реакции, и, соответственно, являющейся методом управления процессом этой реакции. Опытным путем было установлено, что при повышении температуры на каждые 10 0С скорость большинства химических реакций возрастает в 2 – 4 раза (температурный коэффициент скорости реакции). При понижении температуры скорость реакций во столько же раз уменьшается. Значительное увеличение скорости реакций при повышении температуры нельзя объяснить одним только увеличением числа столкновений между молекулами. Согласно кинетической теории, скорость движения молекул растет пропорционально корню квадратному из абсолютной температуры, тогда как скорость реакции увеличивается гораздо быстрее. Следует считать, что повышение температуры не только вызывает более частые столкновения, но и увеличивает число эффективных столкновений, в результате которых происходит химическое взаимодействие, т.е. увеличивает относительное количество активных молекул. Это может быть объяснено тем, что по мере повышения температуры молекулы становятся менее стойкими и, следовательно, более склонными к химической реакции. Для количественного описания температурных эффектов в химической кинетике используют два соотношения - правило Вант-Гоффа и уравнение Аррениуса: Графически зависимость k(T) выглядит следующим образом: 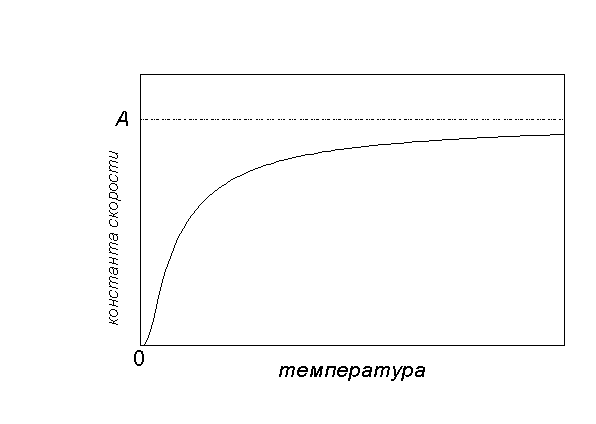 25. Катализ и его особенности Ката́лиз — Явление изменения скорости реакции под воздействием катализаторов. Катализатор — Вещества, не расходующиеся в результате протекания реакции, но влияющие на ее скорость. Реакции, протекающие под действием катализаторов, называются каталитическими. 1. Диффузия реагирующих веществ к поверхности твердого вещества. 2. Физическая адсорбция на активных центрах поверхности твердого вещества реагирующих молекул и затем хемосорбция их. 3. Химическая реакция между реагирующими молекулами. 4. Десорбция продуктов с поверхности катализатора. 5. Диффузия продукта с поверхности катализатора в общий поток. 26. Экспериментальный способ определения порядка и константы скорости реакции. Одной из задач, стоящих перед химической кинетикой, является определение состава реакционной смеси (т.е. концентраций всех реагентов) в любой момент времени, для чего необходимо знать зависимость скорости реакции от концентраций. В общем случае, чем больше концентрации реагирующих веществ, тем больше скорость химической реакции. В основе химической кинетики лежит т. н. основной постулат химической кинетики: Скорость химической реакции прямо пропорциональна произведению концентраций реагирующих веществ, взятых в некоторых степенях. Т. е. для реакции аА + bВ + dD + ... ––> еЕ + ... можно записать: Коэффициент пропорциональности k есть константа скорости химической реакции. Константа скорости численно равна скорости реакции при концентрациях всех реагирующих веществ, равных 1 моль/л. Зависимость скорости реакции от концентраций реагирующих веществ определяется экспериментально и называется кинетическим уравнением химической реакции. Очевидно, что для того, чтобы записать кинетическое уравнение, необходимо экспериментально определить величину константы скорости и показателей степени при концентрациях реагирующих веществ. Показатель степени при концентрации каждого из реагирующих веществ в кинетическом уравнении химической реакции (в уравнении (II.4) соответственно x, y и z) есть частный порядок реакции по данному компоненту. Сумма показателей степени в кинетическом уравнении химической реакции (x + y + z) представляет собой общий порядок реакции. Следует подчеркнуть, что порядок реакции определяется только из экспериментальных данных и не связан со стехиометрическими коэффициентами при реагентах в уравнении реакции. Стехиометрическое уравнение реакции представляет собой уравнение материального баланса и никоим образом не может определять характера протекания этой реакции во времени. Реакции нулевого порядка Для реакций нулевого порядка кинетическое уравнение имеет следующий вид: Скорость реакции нулевого порядка постоянна во времени и не зависит от концентраций реагирующих веществ; это характерно для многих гетерогенных (идущих на поверхности раздела фаз) реакций в том случае, когда скорость диффузии реагентов к поверхности меньше скорости их химического превращения. Реакции первого порядка Рассмотрим зависимость от времени концентрации исходного вещества А для случая реакции первого порядка А ––> В. Реакции первого порядка характеризуются кинетическим уравнением вида: Реакции второго порядка. Для реакций второго порядка кинетическое уравнение имеет следующий вид: 27. Электролиты. Теория электролитической диссоциации С. Аррениуса. Электроли́т — вещество, которое проводит электрический ток вследствие диссоциации на ионы, что происходит в растворах и расплавах, или движения ионов в кристаллических решётках твёрдых электролитов. Примерами электролитов могут служить водные растворы кислот, солей и оснований и некоторые кристаллы, (например, иодид серебра, диоксид циркония). Электролиты — проводники второго рода, вещества, электропроводность которых обусловлена подвижностью ионов.
Электролитическая диссоциация — процесс распада электролита на ионы при его растворении или плавлении. Классическая теория электролитической диссоциации была создана С. Аррениусом и В. Оствальдом в 1887 году. Классическая теория электролитической диссоциации основана на предположении о неполной диссоциации растворённого вещества, характеризуемой степенью диссоциации α, т. е. долей распавшихся молекул электролита. Динамическое равновесие между недиссоциированными молекулами и ионами описывается законом действующих масс . Например, электролитическая диссоциация бинарного электролита KA выражается уравнением типа: Константа диссоциации 28.Теория сильных электролитов. Активность. Коэффициент активности. Зависимость коэффициента активности от ионной силы раствора. Сильные электролиты — химические соединения, молекулы которых в разбавленных растворах практически полностью диссоциированы на ионы. Степень диссоциации таких электролитов близка к 1. К сильным электролитам относятся многие неорганические соли, некоторые неорганические кислоты и основания в водных растворах, а также в растворителях, обладающих высокой диссоциирующей способностью (спирты, амиды и др.). Классическая теория электролитической диссоциации применима лишь к разбавленным растворам слабых электролитов. Сильные электролиты в разбавленных растворах диссоциированы практически полностью, поэтому представления о равновесии между ионами и недиссоциированными молекулами лишено смысла. Согласно представлениям, выдвинутым в 20—30-х гг. 20 в. В. К. Семенченко (СССР), Н. Бьеррумом (Дания), Р. М. Фуоссом (США) и др., в растворах сильных электролитов при средних и высоких концентрациях образуются ионные пары и более сложные агрегаты. Современные спектроскопические данные показывают, что ионная пара состоит из двух ионов противоположного знака, находящихся в контакте («контактная ионная пара») или разделённых одной или несколькими молекулами растворителя («разделённая ионная пара»). Ионные пары электрически нейтральны и не принимают участия в переносе электричества. В сравнительно разбавленных растворах сильных электролитов равновесие между отдельными сольватированными ионами и ионными парами может быть приближённо охарактеризовано, аналогично классической теории электролитической диссоциации, константой диссоциации (или обратной величиной — константой ассоциации). Это позволяет использовать вышеприведённое уравнение для расчёта соответствующей степени диссоциации, исходя из экспериментальных данных. В простейших случаях (большие одноатомные однозарядные ионы) приближённые значения константы диссоциации в разбавленных растворах сильных электролитов можно вычислить теоретически, исходя из представлений о чисто электростатическом взаимодействии между ионами в непрерывной среде — растворителе. Примеры сильных электролитов: некоторые кислоты (HClO4, HMnO4, H2SO4, HCl, HBr; HI), гидроксиды щелочных и щёлочноземельных металлов (NaOH, KOH, Ba(OH)2); большинство солей. Коэффициент активности – коэффициент, связывающий реальную концертрацию электролита с его термодинамической активностью в уравнении a = γc (a – активность; с – концентрация; γ – коэффициент активности). Активность электролита определяется произведением активности его ионов. Так как при бесконечном разбавлении активность каждого из ионов становится тождественной его стехиометрической концентрации, то, предполагая полную диссоциацию и тем самым объясняя аномалии растворов электролита лишь действующими в них силами, мы можем распространить это положение на любой раствор. В бесконечно разбавленных водных растворах неэлектролитов коэффициент активности равен единице. Опыт показывает, что по мере увеличения концентрации электролита величины f уменьшаются, проходят через минимум, а затем снова увеличиваются и становятся существенно большими единицы в крепких растворах. Такой ход зависимости f от концентрации определяется двумя физическими явлениями. Первое особенно сильно проявляется при малых концентрациях и обусловлено электростатическим притяжением между противоположно заряженными ионами. Силы притяжения между ионами преобладают над силами отталкивания, т.е. в растворе устанавливается ближний порядок, при котором каждый ион окружен ионами противоположного знака. Следствием этого является усиление связи с раствором, что находит отражение в уменьшении коэффициента активности. Естественно, что взаимодействие между ионами возрастает при увеличении их зарядов. При возрастании концентрации все большее влияние на активность электролитов оказывает второе явление, которое обусловлено взаимодействием между ионами и молекулами воды (гидратацией). При этом в относительно концентрированных растворах количество воды становится недостаточным для всех ионов и начинается постепенная дегидратация, т.е. связь ионов с раствором уменьшается, следовательно, увеличиваются коэффициенты активности. Известны некоторые закономерности, касающиеся коэффициентов активности. Так, для разбавленных растворов (приблизительно до m = 0,05) соблюдается соотношение 1 - f = k?m. В несколько более разбавленных растворах (т ? 0,01) величины f не зависят от природы ионов. Это обусловлено тем, что ионы находятся на таких расстояниях друг от друга, на которых взаимодействие определяется только их зарядами. При более высоких концентрациях наряду с зарядом на величину активности начинает оказывать влияние и радиус ионов. 29. Слабые электролиты. Константа кислотности и основности. Закон разбалвения Освальда. Слабые электролиты — химические соединения, молекулы которых даже в сильно разбавленных растворах незначительно диссоциированны на ионы, которые находятся в динамическом равновесии с недиссоциированными молекулами. К слабым электролитам относится большинство органических кислот и многие органические основания в водных и неводных растворах. Слабыми электролитами являются:
Константа диссоциации кислоты (Ka) — константа равновесия реакции диссоциации кислоты на ион водорода и анион кислотного остатка. Для многоосновных кислот, диссоциация которых проходит в несколько стадий, оперируют отдельными константами для разных стадий диссоциации, обозначая их как Ka1, Ka2 и т. д. Пример расчета Двухосновной кислоты: Чаще вместо самой константы диссоциации K используют величину pK, которая определяется как отрицательный десятичный логарифм самой константы: Основание — это химическое соединение, способное образовывать ковалентную связь с протоном (основание Брёнстеда ) либо с вакантной орбиталью другого химического соединения (основание Льюиса ). В узком смысле под основаниями понимают основные гидроксиды — сложные вещества, при диссоциации которых в водных растворах отщепляется только один вид анионов — гидроксид-ионы OH-. Теория Брёнстеда — Лоури позволяет количественно оценить силу оснований, то есть их способность отщеплять протон от кислот. Это принято делать при помощи константы основности Kb — константы равновесия реакции основания с кислотой сравнения, в качестве которой выбрана вода. Чем выше константа основности, тем выше сила основания и тем больше его способность отщеплять протон. Часто константу основности выражают в виде показателя константы основности pKb. Например, для аммиака как основания Брёнстеда можно записать: Закон разбавления Оствальда — соотношение, выражающее зависимость эквивалентной электропроводности разбавленного раствора бинарного слабого электролита от концентрации раствора: Здесь К — константа диссоциации электролита, с — концентрация, λ и λ∞ — значения эквивалентной электропроводности соответственно при концентрации с и при бесконечном разбавлении. Соотношение является следствием закона действующих масс и равенства |