реферат. Каталитический крекинг НАЗАРИЙ МАЪЛУМОТЛАР. Каталитический крекинг. Химические основы процесса. Превращения алканов, циклоалканов, алкенов и аренов
 Скачать 1.43 Mb. Скачать 1.43 Mb.
|

Гидрогенизационные процессы. Превращения сероорганических, азотсодержащих, кислородсодержащих и металлоорганических соединений.Гидрогенолиз сероорганических соединений. Меркаптаны гидрируются до сероводорода и соответствующего УВ: Сульфиды гидрируются через образование меркаптанов: Дисульфиды гидрируются аналогично: Циклические сульфиды, например тиофан и тиофен, гидрируются с образованием соответствующих алифатических УВ: Бенз- и дибензтиофены гидрируются по схеме: 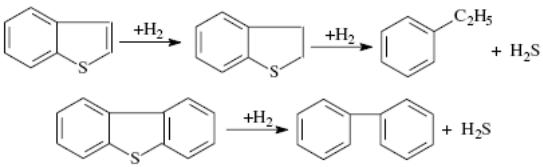 Гидрогенолиз азоторганических соединений. Азот в нефтяном сырье находится преимущественно в гетероциклах в виде производных пиррола и пиридина. Гидрирование их протекает, в общем, аналогично гидрированию сульфидов: 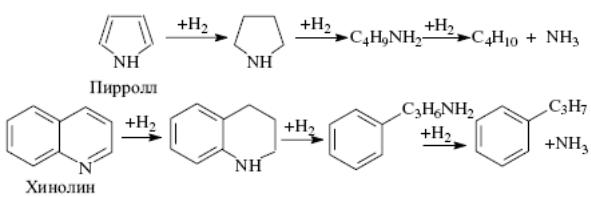 Гидрогенолиз кислородсодержащих соединений. Кислород в топливных фракциях может быть представлен соединениями типа спиртов, эфиров, фенолов и нафтеновых кислот. В газойлевых фракциях и нефтяных остатках кислород находится в основном в мостиковых связях и в циклах полициклических ароматических и смолисто-асфальтеновых соединений нефти. При гидрировании кислородных соединений образуются соответствующие углеводороды и вода: Как видно из термодинамических данных для реакции гидрогенолиза некоторых серо- и кислородсодержащих органических углеводородов, реакции эти экзотермичны и протекают без изменения объема или. в случае гидрогенолиза непредельных гетероорганических соединений (например, производных тиофена), — с уменьшением объема и более высоким экзотермическим эффектом. Следовательно, реакции гидрогенолиза всех гетероорганических соединений являются термодинамически низкотемпературными. Давление не оказывает влияния на равновесие газофазных реакций или благоприятствует образованию продуктов гидрогенолиза. С повышением температуры константы равновесия реакций гидрогенолиза уменьшаются, особенно для тиофена и его производных, но в интервале температур, представляющем практический интерес, равновесие реакций практически нацело смещено вправо для всех гетероорганических соединений, кроме тиофенов, для которых термодинамические ограничения все же ощутимы и их гидрирование проводят при пониженных температурах на высокоактивных катализаторах. На кинетику реакций гидрогенолиза сильное влияние оказывают тип и строение гетероорганичеекнх соединений. Скорость гидрогенолиза в общем возрастает в ряду тиофены<тиофаны<сульфиды<дисульфиды<меркаптаны. С увеличением числа ароматических и циклопарафиновых колец в молекуле сераорганического соединения его реакционная способность относительно гидрогенолиза падает. Так, относительная скорость гидрогенолиза при идентичных условиях для тиофена, бензтиофена и дибензтиофена составляет соответственно 2,9; 2,8 и 1,0. При одинаковом строении реакционная способность относительно гидрогенолиза понижается в ряду гетероорганичеекнх соединений: сераорганические<кислородоорганические<азоторганические. Среди азотсодержащих углеводородов циклические соединения подвергаются гидрогенолизу значительно труднее, чем содержащие азот в аминогруппах. При гидрировании металлорганических соединений образуются металл и соответствующий УВ. Гидрогенизационные процессы. Превращения ув. Катализаторы процесса.Используемые в промышленных гидрогенизационных процессах катализаторы являются сложными композициями, и в их состав входят, как правило, следующие компоненты: - металлы VIII группы: Ni, Со, Pt, Pd. иногда Fe; - окислы или сульфиды VI группы: Mo, W, иногда Сг; - термостойкие носители с развитой удельной поверхностью и высокой механической прочностью, инертные или обладающие кислотными свойствами. Никель, кобальт, платина или палладий придают катализаторам дегидро-гидрирующие свойства, но не обладают устойчивостью по отношению к отравляющему действию контактных ядов и не могут быть использованы в отдельности в гидрогенизационных процессах. Молибден, вольфрам и их оксиды являются n-полупроводниками (как и Ni, Со, Pt и Pd). Их каталитическая активность по отношению к реакциям окисления-восстановления обусловливается наличием на их поверхности свободных электронов, способствующих адсорбции, хемосорбции, гемолитическому распаду органических молекул. Однако Мо и W значительно уступают по дегидро-гидрирующей активности Ni, Со и особенно Pt и Pd. Сульфиды же Мо и W являются р-полупроводниками (дырочными). Дырочная их проводимость обусловливает протекание гетеролитических (ионных) реакций, в частности расщепление C-S, C-N и С-О связей в гетероорганических соединениях. Сочетание Ni или Со с Мо или W придает их смесям и сплавам бифункциональные свойства - способность осуществлять одновременно и гемолитические, и гетеролитические реакции и, что особенно важно, стойкость по отношению к отравляющему действию сернистых и азотистых соединений, содержащихся в нефтяном сырье. Применение носителей позволяет снизить содержание активных компонентов в катализаторах, что особенно важно в случае использования дорогостоящих металлов. В зависимости от типа реакторов катализаторы на носителях изготавливают в виде таблеток, шариков или микросфер. Носители нейтральной природы (оксиды алюминия, кремния, магния и др.) не придают катализаторам на их основе дополнительных каталитических свойств. Носители, обладающие кислотными свойствами, как, например, синтетические аморфные и кристаллические алюмосиликаты и цеолиты, магний- и цирконийсиликаты. фосфаты, придают катализаторам дополнительно изомеризующие и расщепляющие (крекирующие) свойства. Отсюда понятно, почему катализаторы гидрообессеривания высококипящих и остаточных нефтяных фракций, особенно гидрокрекинга, изготавливают с использованием кислотно-активных носителей. Катализаторы на таковых носителях, содержащие металлы VI и VIII групп, являются по существу полифункциональными. В мировой практике наибольшее распространение в гидрогенизационных процессах получили алюмокобальтмолибденовые (АКМ), алюмоникельмолибденовые (АНМ) и смешанные алюмоникелькобальтмолибденовые (АНКМ), а также алюмоникельмолибденсиликатные (АНМС) катализаторы. В процессах глубокого гидрирования азотсодержащих и ароматических соединений парафинов и масляных фракций применяют алюмоникель- или алюмокобальтвольфрамовые катализаторы (АНВ или АКВ). В последние годы распространение получают цеолитсодержащие катализаторы гидрообессеривания и гидрокрекинга. АКМ и АНМ катализаторы гидроочистки содержат 2..4% мас. Со или Ni и 9...15% мас. МоО3 на активном γ-оксиде алюминия. На стадии пусковых операции или в начале сырьевого цикла их подвергают сульфидированию (осернению) в токе H2S и Н2, при этом их каталитическая активность существенно возрастает. Активность АКМ и АНМ катализаторов зависит как от суммарного содержания в них гидрирующих компонентов (Со+Мо или Ni+Мо), так и ототношения Со/Со + Мо и Ni/Ni + Мо. У большинства марок зарубежных катализаторов гидроочистки и гидрообессеривания суммарное содержание гидрирующих компонентов составляет 16-21% мас, а отношение Co(Ni)/Co(Ni) + Мо колеблется в пределах 0.17...0,28. У отечественных катализаторов АКМ, АНМ и АНМС эти показатели составляют соответственно 16 и 0,52. АКМ катализатор высокоактивен в реакциях гидрогенолиза сернистых соединений и обладает достаточно высокой термостойкостью. Он достаточно активен в реакциях гидрирования непредельных углеводородов, азотистых и кислородсодержащих соединений сырья и применим для гидроочистки всех топливных фракций нефти. Однако большой дефицит кобальта ограничивает его распространение. АНМ катализатор, по сравнению с АКМ, более активен в реакциях гидрирования ароматических углеводородов и азотистых соединений и менее активен в реакциях насыщения непредельных соединений. Однако у него несколько ниже показатели по термостойкости и механической прочности. АНМС катализатор имеет тот же состав гидрирующих компонентов, что и АНМ. Изготавливается добавлением к носителю (γ-оксиду алюминия) 5...7% мас. диоксида кремния. При этом увеличивается его механическая прочность и термостойкость, незначительно улучшается гидрирующая активность. Катализаторы ГО-30-70 и ГО-117 отличаются от вышерассмотренных большим содержанием гидрирующих компонентов (до 28% мас), несколько большей каталитической активностью и повышенной механической прочностью. Катализаторы ГS-168ш и ГК-35 промотированы введением в состав их носителей соответственно алюмосиликата и цеолита типа Y и потому обладают повышенной расщепляющей активностью: могут использоваться для гидрооблагораживания дизельных и газойлевых фракций, а также гидрокрекинга дистиллятного сырья. Катализатор ГКД-202 отличается от ГК-35 меньшим содержанием гидрирующих металлов (18% мас); изготавливается с использованием в качестве носителя алюмосиликата с добавкой цеолита; обладает наилучшими показателями по механической прочности, межрегенерационному пробегу и сроку службы катализатора; по активности в реакциях обессеривания находится на уровне катализаторов АКМ и АНМ. Этот катализатор является базовым для процессов гидроочистки реактивных и дизельных фракций — сырья процессов цеолитной депарафинизации. Несмотря на проведенные во многих странах мира многолетние исследования с применением комплекса разнообразных физико-химических методов до сих пор не установлено, какие именно структуры и фазовый состав катализаторов гидрогенизационных процессов соответствуют каталитически активному их состоянию. Кобальт (никель) и молибден (вольфрам) образуют между собой сложные объемные и поверхностные соединения типа молибдатов (вольфраматов) кобальта (никеля), которые при сульфировании формируют каталитически активные структуры сульфидного типа СоxMoSy (NixMoSy, СоxWSy, NixWSy). Возможно также образование на поверхности носителя Аl2O3, каталитически неактивных мнительных фаз типа алюминатов кобальта (никеля) и молибдата (вольфрамата) алюминия. Наиболее вероятной структурой в сульфидированных АКМ катализаторах, ответственной за бифункциональные их каталитические свойства, считается фаза CoMoS2. По аналогии с механизмами реакций, осуществляемых в процессах каталитического риформинга на платине и паровой конверсии углеводородов, можно предположить, что реакции гидрогенолиза гетероатомных углеводородов на АКМ и АНМ катализаторах протекают также многостадийно через хемосорбцию реактантов на активных центрах как кобальта (никеля), так и молибдена. При этом на кобальте (никеле) осуществляются активация Н2 и спилловер атомарного активного водорода, а на молибдене протекают сульфирование (осернение), азотирование и окисление с образованием поверхностных соединений Mo(S), Mo(N) и Мо(О), которые под действием активированного водорода подвергаются десульфированию (обессериванию), деазотированию и восстановлению: 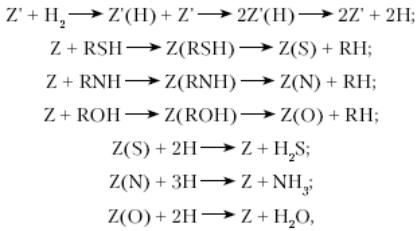 где Z' и Z – соответственно активные центры кобальта (никеля) и молибдена. При установившемся режиме в процессе достигается стационарное состояние по поверхностным концентрациям σS, σN и σO в зависимости от прочности связей C-S, C-N и С-О, активности катализатора и параметров гидрогенолиза. При этом активные центры кобальта (никеля) при избытке водорода полностью заняты активированным водородом (отсюда серостойкость катализаторов и кажущийся нулевой порядок суммарной реакции но водороду). Возможны также иные маршруты элементарных реакций гидрогенолиза, в том числе через мультиплетную хемосорбцию реактантов, что энергетически более выгодно. |