реферат. Каталитический крекинг НАЗАРИЙ МАЪЛУМОТЛАР. Каталитический крекинг. Химические основы процесса. Превращения алканов, циклоалканов, алкенов и аренов
 Скачать 1.43 Mb. Скачать 1.43 Mb.
|

Каталитический крекинг алкеновАлкены не содержатся в нефтяных фракциях, но образуются при термическом разложении алканов и циклоалканов, и их термокаталитические превращения определяют состав конечных продуктов процесса. Поэтому закономерности каталитических превращений алкенов в условиях каталитического крекинга представляют особый интерес. Скорость каталитического крекинга алкенов на два-три порядка выше скорости крекинга соответствующих алканов, что объясняется легкостью образования из алкенов карбениевых ионов: При присоединении протона к молекуле алкена образуется такой же ион, как и при отщеплении гидрид-иона от алкана, что определяет общность их реакций при каталитическом крекинге. Кроме образования низших алканов и алкенов каталитический крекинг алкенов приводит к образованию циклоалканов и аренов. Механизм этих процессов может быть представлен схемой: 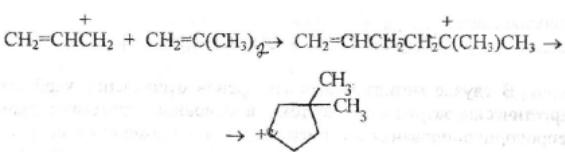 Далее может произойти изомеризация в шестичленный цикл и превращение в арен. Каталитический крекинг алкилароматических углеводородовНезамещенные арены в условиях каталитическою крекинга устойчивы. Метилзамещенные арены реагируют со скоростью, близкой к алканам. Алкилпроизводные аренов, содержащие два и более атомов углерода в цепи, крекируются примерно с такой же скоростью, что и алкены. При крекинге алкилароматических углеводородов бензольное кольцо не затрагивается, тогда как боковые цепи во всех случаях, кроме толуола, отщепляются с образованием олефина. Влияние длины цепи и ее разветвленное на энергию активации (в кДж/моль) показано ниже:  Обычно скорость крекинга боковых цепей возрастает при переходе от первичного к вторичному и третичному углеродным атомам, соединяющим цепь с кольцом. Для одного и того же типа присоединения скорость растет при увеличении длины боковой цепи. 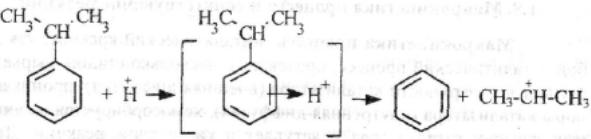 В случае метилзамещенных аренов отщепление карбкатиона энергетически затруднено, поэтому в основном протекают реакции диспропорционирования и изомеризации по положению заместителей. В случае толуола доминирующей реакцией является диспропорционирование в бензол и ксилол, а не крекинге отщеплением метана. Полиметилбензолы претерпевают главным образом изомеризацию и диспропорционирование. Например, ксилолы в присутствии кислотных катализаторов изомеризуются и диспропорцинируются в соответствии со следующей схемой: 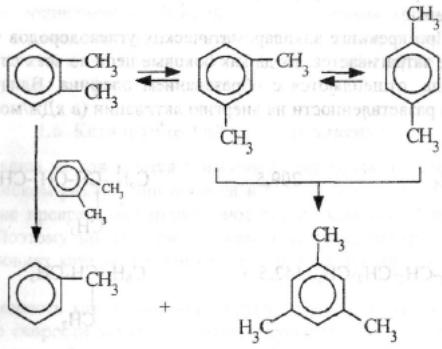 Полициклические арены прочно сорбируются на катализаторе и подвергаются постепенной деструкции и перераспределению водорода с образованием кокса. 18. Каталитический крекинг. Принципиальная технологическая схема. Режим процесса.Чаще всего сырьем для каталитического крекинга является широкие вакуумные фракции (350-500оС). Кроме того, каталитическому крекингу можно подвергать сырье вторичного происхождения: газойли коксования, термического крекинга и гидрокрекинга. На рис. 7 приведена принципиальная технологическая схема установки каталитического крекинга Г-43-107. производительность по сырью 160 т/ч. 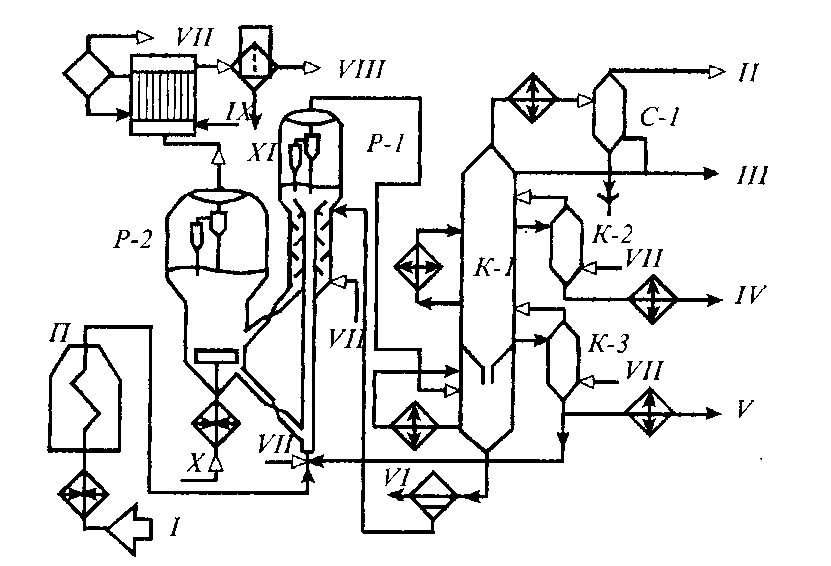 Рисунок 7. Принципиальная технологическая схема установки КК. I— гидроочищенное сырье; II — газы на ГФУ; III — нестабильный бензин; IV— легкий газойль; V — тяжелый газойль; VI — декантат; VII — водяной пар; VIII— дымовые газы; IX — вода; X — воздух; XI — катализаторная пыль. Гидроочищенное сырье нагревается в теплообменниках и печи (до 340оС) смешивают с рециркулятом и водяным паром и направляют в узел смешения прямоточного лифт-реактора Р-1. При контакте с регенерированным горячим цеолитсодержащим катализатором сырье испаряется и подвергается каталитическому крекингу в лифт реакторе (при температуре 540-560оС). Затем сырье с катализатором поступает в зону форсированного кипящего слоя в реактор Р-1. Продукты реакции отделяют от катализаторной пыли в двухступенчатых циклонах и направляют в нижнюю часть ректификационной колонны К-1 на дальнейшее разделение. Отработанный, закоксованный катализатор (с содержанием кокса 0.5-0.6%) из отпарной зоны Р-1 по наклонному катализаторопроводу направляют в зону кипящего слоя регенератора Р-2, где происходит выжиг кокса в режиме полного окисления оксида углерода (при температуре 640-650оС). Затем регенерированный катализатор (с содержанием кокса 0.005-0.1%) по нижнему наклонному катализаторопроводу поступает в узел смешения лифт-реактора. Воздух для регенерации катализатора нагнетают воздуходувкой в реактор Р-2. При этом дымовые газы проходят через внутренние двухступенчатые циклоны и затем их направляют на утилизацию теплоты в котел-утилизатор и перед сбросов в атмосферу очищают от пыли на электрофильтрах. В колонне К-1 предусмотрено верхнее (острое) и промежуточное циркуляционное орошение (в средней и нижней части колонны). Легкий и тяжелый газойль отбирают через отпарные колонны К-2 и К-3. При этом нижняя часть колонны играет роль скруббера для каталитического шлама, который возвращают в отпарную зону реактора Р-1. В качестве рециркулята часть тяжелого газойля подают в узел смешения лифт-реактора. Смесь паров бензина, воды и газов выводят с верха колонны. Затем эту смесь охлаждают в холодильнике и разделяют в газосепараторе С-1. Газы направляют на установку ГФУ, нестабильный бензин на стабилизацию, а водный конденсат после очистки от сернистых соединений выводят с установки. Таким образом, в результате каталитического крекинга фракции 350-500оС (содержание серы 0.2%)получается, % мас.: 1.96 – сухой газ; 5.61 – пропан-пропиленовая фракция; 9.04 – бутан-бутиленовая фракция; 43.04 – бензиновая фракция (С5- 195оС); 28.0 – легкий газойль (дизельная фракция 195-350оС); 8.35 – тяжелый газойль (более 350оС); 4 – кокс + потери. Каталитический крекинг кумола ⇐ Предыдущая16171819202122232425Следующая ⇒ Каталитический крекинг углеводородов нефти является одни из основных промышленных процессов нефтепереработки. Впервые превращения углеводородов в присутствииалюмосиликатов были изучены Л.Г. Гурвичем и С.В. Лебедевым. В настоящее время химизм и механизм превращений углеводородов над алюмосиликатными катализаторами исследованы в значительной степени. Установлено, что механизм превращения углеводородов в присутствии алюмосиликатных катализаторов носит ионный характер. В условиях каталитического крекинга углеводороды подвергаются сложным превращениям, а именно реакциям расщепления,изомеризации, дегидрирования, перераспределения водорода. Для алкилбензолов характерным является расщепление алкильной цепи по α-связи С-С с образованием бензола и олефина. Например, в случае изопропилбензола: 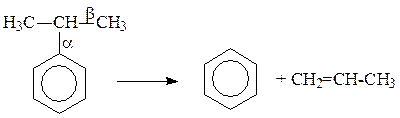 Расщепление наиболее прочной α-связи С-С при каталитическом крекинге алкилбензолов (при термическом крекинге расщепляете наиболее слабая β-связь) обусловливается сильной поляризацией α-связи в промежуточном комплексе с катализаторомс ее последующимгетеролитическим расщеплением: 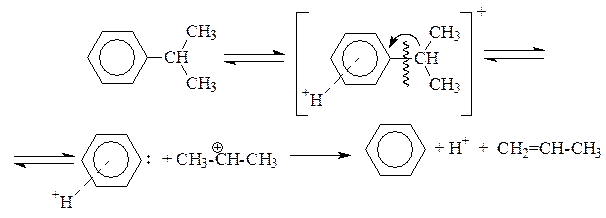 В последнее время в качестве возможного рассматривается катион-радикальныймеханизм каталитического крекинга.  При взаимодействии изопропилбензола с активным (льюисовским) центром катализатора происходит переход одного π-электрона на вакантную d-орбиталь атома алюминия в алюмокислородном тетраэдре (А1О4) с образованием катион-радикала углеводорода, находящегося в комплексе с анион-радикалом активного центра. Далее происходит распад комплекса с отщеплением изопропила и фенильного катиона сорбированного анион-радикалом катализатора. Последний отдает π-электрон фенильному катиону, который превращается в фенил и процесс продолжается по радикальному механизму. Таким образом, основными продуктами крекинга кумола над алюмосиликатным катализатором являются бензол и пропилен. В качестве побочных продуктов образуются "кокс", а также толуол, этилбензол, метан, водород и другие газообразные вещества. Образование побочных продуктов усиливается за счет реакции перераспределения водорода, а также вследствие деактивации катализатора (блокирование активных центров катализатора углистыми отложениям - "коксом"). В последнем случае, по-видимому, начинает становиться заметным превращение кумола только по радикальному механизму (на поверхности частиц "кокса"), что объясняет присутствие в газообразных продуктах толуола, этилбензола, метана и водорода: 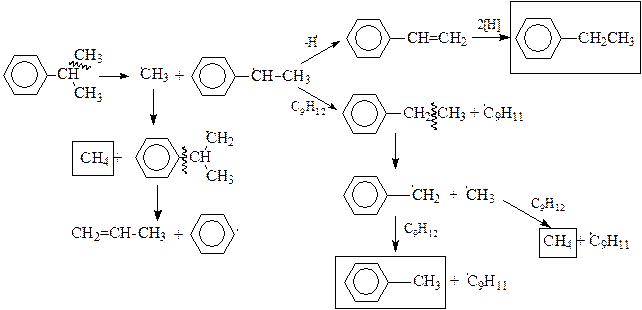 Цель работы стоит в определении химизма крекинга кумола в заданных условиях. Проведение опыта и обработка результатов Опыт ведется на установке для изучения каталитических реакций в потоке (рис. 12). 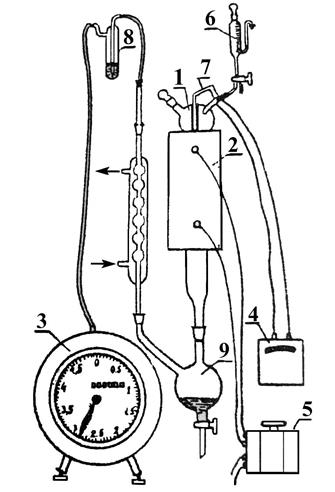 Рис. 12. Прибор для проведения каталитических реакций в потоке: 1 – реактор, 2 – электрическая печь, 3 – газовые часы, 4 – милливольтметр, 5 – ЛАТР, 6 – бюретка, 7 – термопара, 8 – счетчик пузырьков, 9 – приемник катализата В реактор – кварцевую трубку – помещают 70млалюмосиликатного катализатора (катализатор предварительно обезвоживают в муфельной печи при 500°С в течение двух часов) и засыпают кварцевую насадку для обеспечения равномерного нагрева и испарения сырья. Включают электрообогрев печи. Температуру в печи измеряют хромель-алюмелевой термопарой по милливольтметру. Регулирование температуры осуществляется латром. После установления в печи температуры в пределах 450-500°С (по заданию преподавателя) приступают к опыту. В бюретку наливают 10 мл (9,3 г) кумола, устанавливают объемную скорость (V) (объемная скорость - это отношение объема сырья, подаваемого в реактор, в час к объему подачи кумола) в пределах 0,4-1 ч-1 (по заданию преподавателя). Подача кумола в реактор может проводиться с помощью специального дозатора. Скорость подачи кумола (в см/ч) в реактор находят по формуле: υ=Vк-V, где Vк – объем слоя катализатора, см3; V - объемная скорость подачи, ч-1. Устанавливают скорость подачи кумола в реактор по числу капель кумола, поступающего в реактор в минуту (предварительно необходимо определить число капель в 1 см3 кумола). После установления скорости записывают время начала опыта, а катализат, образовавшийся при установлении скорости, сливают и выливают в сливную емкость. Жидкие продукты крекинга собираются в приемнике, а газы через холодильник поступают в вытяжное устройство. Объем выделившегося газа определяют следующим образом. Газ поступает в перевернутый цилиндр, заполненный водой и находящийся в водяной бане. По мере поступления газ вытесняет воду. Определяется объем газа, выделившегося в единицу времени (минуту). По окончании подачи кумола записывают время окончания опыта и рассчитывают фактический объем газа за время опыта; выключают обогрев печи и в течение 5 минут дают возможность продуктам реакции стечь из реактора в приемник. Жидкие продукты крекинга (катализат) взвешивают, записывают объем газа Материальный баланс крекинга составляют по форме: Материальный баланс крекинга.
Катализат анализируют методом газовой хроматографии на хроматографе с применением капиллярной колонки длиной 50 м, диаметром – 0,2 мм с неподвижной жидкой фазой трикрезилфосфат. Детектор – ДИП. Условия анализа: температура в колонке 110°С, в испарителе 250°С, в детекторе 150°С, газ-носитель – гелий. После достижения прямой нулевой линии на дисплее (через прибор пропускается только гелий), вводят пробу катализата в количестве 0,02 - 0,04 мкл. Хроматограмма катализата обычно имеет вид (рис. 13): 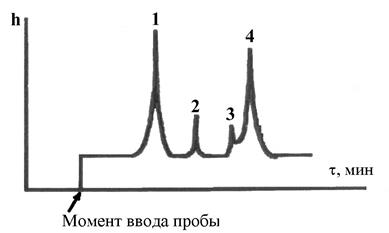 Рис. 13. Хроматограмма фракции: 1-бензол; 2-толуол; 3-этилбензол; 4-кумол. Хроматограмму рассчитывают методом внутренней нормализации. Определяют концентрацию кумола во фракции 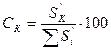 где Зная массу катализата и содержание в нем кумола, находят количество непревращенного кумола. Рассчитывают конверсию кумола: где КВ - масса кумола, взятого для крекинга; КН - масса непревращенного кумола. На основании этих данных и данных анализа газа делают выводы о химизме крекинга. 1. Во избежание ожогов необходимо соблюдать осторожность при работе с трубчатой печью, температура в печи 500 °C. 2. Нельзя трогать провода, подводящие ток к печи и к автотрансформатору, и пытаться самостоятельно без помощи лаборанта что-либо делать с электрической проводкой. 3. Перед подачей углеводорода в реактор необходимо убедиться в том, что кран бюретки не подтекает и пробка крана закреплена с помощью резинового жгута. 4. Перегонку продуктов крекинга необходимо проводить в вытяжном шкафу. Колбу необходимо нагревать с помощью закрытой электрической плитки. План отчета 1. Введение. Цель работы. 2. Описание опыта и его материальный баланс. 3. Разгонка и материальный баланс разгонки. 4. Анализ продуктов крекинга. 5. Расчет конверсии кумола. 6. Выводы о химизме крекинга. Контрольные вопросы 1. Каков химизм и механизм крекинга алкилбензолов над алюмосиликатным катализатором? 2. Каково влияние углистых отложений («кокса») на химизм и механизм каталитического крекинга углеводородов? 3. Расскажите о природе активных центров алюмосиликатных катализаторов. 4. Какие углеводороды могут находиться в промежуточной фракции (95-140°С) катализата? В остатке? Каков химизм и механизм их образования? Дегидрирование циклогексана в присутствии Опыты проводят на установке для исследования каталитических реакций в потоке (рис. 12). В реактор, содержащий 70 мл катализатора, при температуре 520-530°С подают с объемной скоростью 0,2-0,3 час-1 циклогексан. Предварительно устанавливают скорость подачи циклогексана (капли в минуту) в соответствии с заданной преподавателем объемной скоростью. После установления скорости подачи циклогексана из приемника сливают катализат, образовавшийся во время установления скорости подачи, и с этого момента фиксируют, записывают и начинают отсчет времени опыта. В реактор подают 5 мл циклогексана, поддерживая постоянными температуру и скорость подачи циклогексана. Во время опыта измеряют объем выделившегося за какой-либо отрезок времени газа (водород), собирая его в градуированный цилиндр над слоем воды. По окончании опыта записывают время его проведения и рассчитывают фактическую объемную скорость подачи циклогексана по формуле: где υ скорость подачи циклогексана, мл/час. Взвешивают катализат, определяют его показатель преломления при температуре его определения (t), а затем пересчитывают на 20°с по формуле: На основании где Составляют материальный баланс опыта по форме:
Примечание: плотность циклогексана Сравнивают конверсию циклогексана, полученную экспериментально, с теоретической конверсией. Для определения теоретической конверсии вначале определяют величину энергии Гиббса реакции дегидрированияциклогексана по формуле: ∆G = 53800 -96.6 Т Затем вычисляют логарифм константы равновесия: и потаблице 10определяют теоретическую конверсию. Таблица 10
Сравнивают теоретическую конверсию с экспериментальной и делают выводы. 1. Необходимо соблюдать осторожность при работе с трубчатой печью, температура в печи около 350°С. 2. Нельзя трогать провода, подводящие ток к печи и к автотрансформатору и пытаться самостоятельно без помощи лаборанта что-нибудь исправлять в электропроводке во время работы прибора. План отчета 1. Введение. Цель работы. 2. Описание опыта, материальный баланс. 3. Исследование катализата. Определение процентного содержания бензола в катализате и определение конверсии циклогексана в бензол. 4. Расчет теоретической конверсии циклогексана. 5. Выводы. Контрольные вопросы 1. Какие реакции могут протекать с углеводородам других рядов при дегидрировании циклогексана и его гомологов над Pt, Pd, Ni? 2. Выше какой температуры свободная энергия реакции дегидрирования циклогексана становится отрицательной? 3. Какие гомологи циклогексана не способны дегидрироваться в условиях реакции Зелинского? 4. Каковы современные представления о механизме дегидрирования циклогексана над металлическими катализаторами? 5. Каково аналитическое значение реакции Зелинского? 2.3. Дегидроциклизация н-гептана над оксидными катализаторами Реакция дегидроциклизации н-алканов была открыта советскими учеными в 1936-1937 гг. При пропускании алканов состава С6-С7 над окисью хрома или сульфидом молибдена при температурах 450-470°C Б.Л. Молдавскийи Г.Д. Камушер получили ароматические углеводороды. При этом в молекулах ароматических углеводородов содержалось то же количество атомов углерода, что и в молекулах исходных углеводородов. Так,из н-гептана был получен толуол; из 2,5-диметилгексана – п-ксилол. Б. А. Казанский, А.Ф. Платэ осуществили реакцию дегидроциклизации алканов В присутствии катализатора платина на угле при 300-310°С, В.И. Каржев, М.Г. Северьянова и А.Н. Сиова – в присутствии медно-хромового катализатора. В настоящее время в этой реакции изучены многие индивидуальные углеводороды и различные катализаторы. Реакция дегидроциклизации алканов играет большую роль в процессах каталитического риформинга углеводородов нефти Во всех нефтях, помимо углеводородов, содержатся соединения, имеющие в молекуле гетероатомы, такие как сера, кислород, азот. Содержание серы в нефтях обычно колеблется от сотых долей до 8%. Редко может быть и выше. По содержанию серы нефти подразделяют на: 1 – малосернистые до 0,6%; 2 – сернистые от 0,61 до 1,8%; 3 – высокосернистые от 1,81 до 3,5%; 4 – особо высокосернистые свыше 3,5%. В настоящее время более 60% разрабатываемых мировых запасов нефтей являются сернистыми и высокосернистыми. Сернистые соединения считаются вредными компонентами нефтей. Они осложняют процессы переработки, транспортировки и добычи нефтей, вызывая коррозию нефтяного оборудования, дезактивируя катализаторы, загрязняя атмосферу выделяющимися токсичными газами (H2S, SO2, CH3SH). Кроме того, наличие сернистых соединений в нефтепродуктах ухудшает их эксплуатационные характеристики. Метод качественного определения серы в нефтепродуктах называется “проба на медную пластинку”. Метод заключается в том, что отполированную медную пластинку выдерживают в нефтепродукте 12 часов при 85°С. Если медная пластинка покрывается черным налетом, это является признаком наличия серы в нефтепродукте. О количестве сернистых соединений в нефтях и нефтепродуктах судят по результатам определения общей серы. Существует несколько методов определения общей серы. Все они основаны на сожжении навески нефтепродукта и дальнейшем количественном определении образующегося оксида серы (IV). К таким методам относятся: сожжение в трубке ГОСТ 1437-75, ламповый метод. Первый метод предназначен для определения серы в темных нефтепродуктах. Ламповый метод применяется для определения серы в светлых нефтепродуктах. Ламповый метод наиболее прост в выполнении. Однако точность этого метода понижается с повышением температуры кипения исследуемой фракции вследствие неполного сгорания высокомолекулярных соединений, о чем свидетельствует образование копоти и нагара на фитиле. Наиболее удачной модификацией лампового метода, позволяющей определять серу как в светлых, так и в темных нефтепродуктах и сырых нефтях, является так называемый «пиролитический ламповый метод» (он же метод двойного сожжения). Пиролитический ламповый метод заключается в том, что навеску анализируемого вещества испаряют и пиролизуют в кварцевой ампулке, открытый конец которой введен в дожигающее пламя диоксановой горелки (рис.9). Продукты испарения и разложения полностью сгорают в дожигающем пламени, а образующиеся при этом оксид серы (IV) и оксид углерода (IV) улавливаются поглотительным титрованным раствором карбоната натрия. Уменьшение титра поглотительного раствора определяется последующим титрованием его кислотой. Титрование раствора соды проводят 0,05н или 0,1н раствором НСl или Н2SО4 до перехода окраски раствора от зеленой до красно-фиолетовой (в качестве индикатора добавляют 2-3 капли метилового оранжевого и 4-5 капель индигокармина). 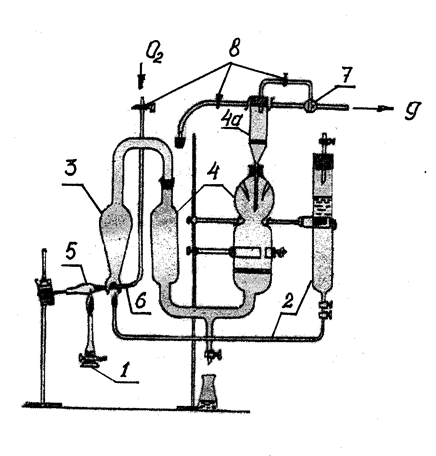 Рис.9. 1– газовая горелка Теклю с шипящим пламенем; 2– диоксановая горелка с двумя кранами; 3– ламповое кварцевое стекло; 4– поглотительный сосуд, состоящий из абсорбера 4 и туманоуловителя 4а; 5– кварцевый стаканчик для сожжения навески вещества; 6– кварцевый капилляр; 7– тройник; 8– винтовые зажимы (3 шт.); 9– к вакуум-насосу для всасывания воздуха и продуктов сгорания. Процентное содержание серы в исследуемом продукте вычисляют по формуле: 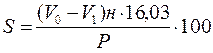 , ,где V0 – количество (мл) раствора кислоты, пошедшее на титрование раствора, полученного в холостом опыте; V1 – количество (мл) раствора кислоты, пошедшее на титрование раствора, полученного в опыте с навеской; н – нормальность раствора кислоты; Р – навеска, мг; 16,03 – эквивалентная масса серы. Метод двойного сожжения позволяет легко и достаточно точно определить количественное содержание серы в различных органических соединениях и нефтепродуктах, а также в нефтях. |