реферат. Каталитический крекинг НАЗАРИЙ МАЪЛУМОТЛАР. Каталитический крекинг. Химические основы процесса. Превращения алканов, циклоалканов, алкенов и аренов
 Скачать 1.43 Mb. Скачать 1.43 Mb.
|

Катализаторы процесса каталитического крекингаПриведенное выше рассмотрение механизма процесса каталитического крекинга основывалось на том, что химические превращения инициировались протоном. Таким образом, каталитический крекинг является процессом, который катализируется кислотами Бренстеда-Лаури. Казалось бы, что в каталитическом крекинге в качестве катализаторов можно использовать обычные минеральные кислоты, например, серную, фосфорную и т.д. Подобного типа минеральные кислоты действительно способны привести к расщеплению высших углеводородов на низшие алканы и алкены. Однако в промышленной органической химии они не могут быть использованы. Связано это с тем, что жидкие кислоты (даже слабые карбоновые кислоты) при высоких температурах вызывают интенсивную коррозию металлической аппаратуры. Поэтому в каталитическом крекинге практическое применение нашли твердые кислоты, которые в минимальной степени способны взаимодействовать со стенками аппаратуры. Использование твердых кислот для катализа предполагает, что углеводороды из газовой фазы первоначально должны адсорбироваться на поверхности твердой фазы. Затем, в ходе миграции адсорбированной молекулы углеводорода по поверхности твердой фазы, она должна встретиться с кислотным центром. На этом центре должны пройти процессы распада молекулы высшего углеводорода. В конце процесса образовавшиеся молекулы продуктов реакции с поверхности твердой фазы должны десорбироваться в газовую фазу. Естественно, поскольку процессы каталитического крекинга протекают на поверхности твердого катализатора, то чем больше площадь этой поверхности, то тем выше должна быть активность катализатора. Поэтому катализаторы, используемые в каталитическом крекинге, должны обладать высокой пористостью. В качестве таких материалов наиболее подходящими оказались цеолиты. Краткие сведения о цеолитах. Цеолиты (буквальный перевод с греческого – кипящий камень) – это неорганические полимеры, получаемые поликонденсацией орто-кремниевой кислоты и гидроокиси алюминия. Эти полимеры относятся к классу алюмосиликатов. Алюмосиликаты составляют основу природных глин. Название цеолитов отражает тот факт, что они являются высокопористыми веществами. 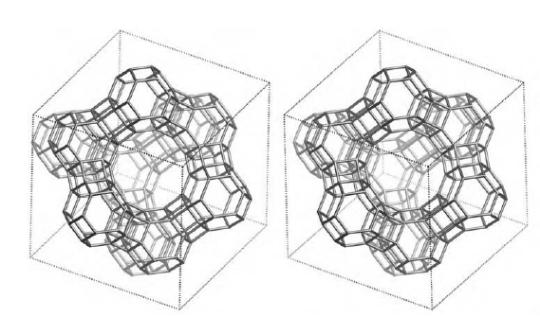 Рис. 1. Структура алюмосиликата типа “Цеолит Y”. Показаны виды спереди и сзади. 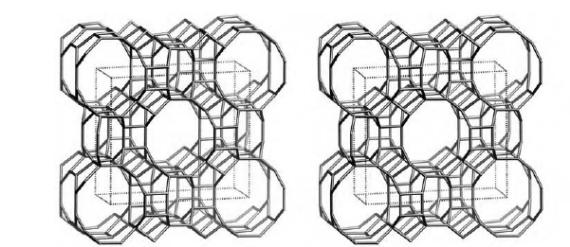 Рис. 2. Структура алюмосиликата типа “Морденит”. 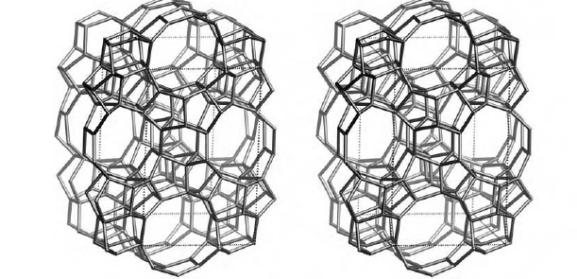 Рис.3. Структура алюмосиликата типа “ZSM-5”. Структура алюмосиликатов значительно зависит от соотношения алюминия и кремния в этих материалах. В зависимости от этого соотношения образуются алюмосиликаты, обладающие разной кристаллической структурой. На рис. 1-3 в качестве примера приведены структуры некоторых алюмосиликатов. Приведенные выше данные по структурам цеолитов указывают на то, что в них существуют поры. Благодаря этому цеолиты обладают высокой удельной поверхностью (отношение общей поверхности пористого тела к его объему или массе). У цеолитов удельная поверхность достигает величины более 1000 м2/г. Исторически применение алюмосиликатов в процессах каталитического крекинга прошло ряд этапов. Первоначально в качестве катализаторов в этих процессах использовались природные алюмосиликаты – глины. Использование природных алюмосиликатов приводило к повышенной степени образования кокса в ходе крекинга. Оказалось, что к этому крайне нежелательному процессу приводят соединения тяжелых металлов (железа, никеля, кобальта, олова и т.д.), которые всегда в незначительных количествах присутствуют в природных алюмосиликатах. Поэтому встала задача получения алюмосиликатов, которые не содержали бы следов тяжелых металлов. Путь решения этой задачи потребовал организации получения синтетических алюмосиликатов. При получении синтетических алюмосиликатов используют очищенные до высокой степени гидроокись алюминия и ортокремневую кислоту. Тем самым исключается попадание в алюмосиликаты соединений тяжелых металлов. На первых этапах развития промышленности синтетических алюмосиликатов производились аморфные алюмосиликаты. Аморфные алюмосиликаты не обладали упорядоченной структурой. Их структура представляла беспорядочную трехмерную сеть из взаимосвязанных оксидов кремния и алюминия. Они обладали слабо развитой поверхностью. Поэтому аморфные алюмосиликаты обладали пониженной каталитической активностью. В дальнейшем было освоено производство высокопористых кристаллических алюмосиликатов (структуры некоторых кристаллических алюмосиликатов приведены выше), которые применяются в качестве катализаторов в настоящее время. Причина появления каталитической активности у алюмосиликатов в процессах каталитического крекинга. Приведенное выше рассмотрение механизма каталитического крекинга указывает на то, что необходимым условием для осуществления процессов является присутствие в системе кислот Бренстеда-Лаури. Поверхностный слой алюмосиликатов содержит гидроксильные группы. Поэтому можно было бы полагать, что эти гидроксильные группы и являются источником протонов. Однако оказалось, что сами алюмосиликаты не обладают никакой каталитической активностью в процессах каталитического крекинга. Алюмосиликаты начинают катализировать эти процессы только тогда, когда они предварительно проходят высокотемпературную обработку (8000С и выше). Если прокаленные алюмосиликаты выдержать во влажной атмосфере, то их каталитическая активность полностью исчезает, но она вновь появляется при их повторном прокаливании. Эти экспериментальные данные указывают на то, что не поверхностные гидроксильные группы в цеолитах являются ответственными за явление катализа в обсуждаемом процессе. При прокаливании алюмосиликатов в них протекают какие-то превращения, которые и являются ответственными за факт появления у них каталитической активности. Выяснение причин появления каталитической активности у алюмосиликатов потребовало длительных исследований. Полное решение этой проблемы стало возможным только тогда, когда появились прецизионные методы определения содержания гидроксильных групп в них, и метода ядерного магнитного резонанса высокого разрешения в твердом теле. Последний метод позволял судить об электронном состоянии атомов в алюмосиликатах. Родственными алюмосиликатам по химической природе являются различные модификации двуокиси кремния. Она также является неорганическим полимером. Поверхностный слой двуокиси кремния также содержит гидроксильные группы. Эти гидроксильные группы легко регистрируются методом ИК-спектроскопии. Оказалось, что при прокаливании различных модификаций двуокиси кремния происходит полная потеря гидроксильных групп. Прокаленная двуокись кремния не обладает никакой каталитической активностью в процессах каталитического крекинга. Наблюдаемое явление указывает на то, что при термической обработке происходит дегидратация поверхностного слоя двуокиси кремния: 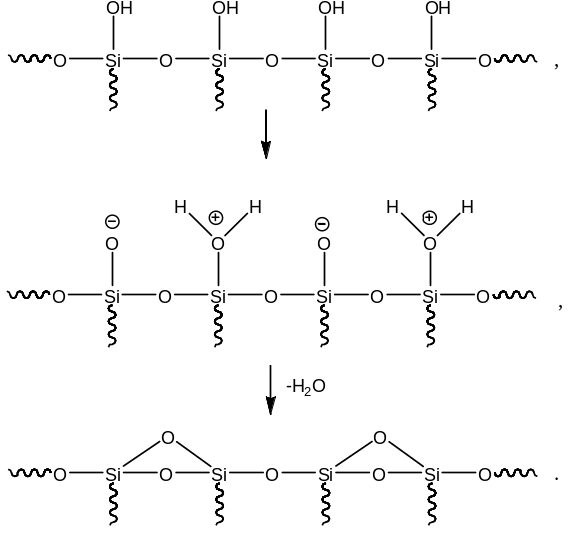 В алюмосиликатах поверхностный слой также содержит гидроксильные группы при атомах кремния. Однако в этом случае гидроксильные группы пространственно разделены по сравнению с двуокисью кремния. Ответственными за это являются атомы алюминия, включенные в структуру полимерной цепи: 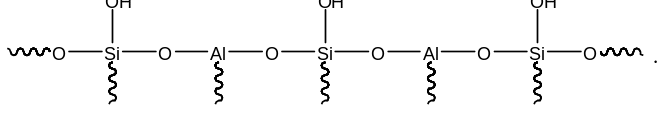 При термической обработке алюмосиликатов, как в случае двуокиси кремния происходит дегидратация за счет отщепления молекул воды при взаимодействии поверхностных гидроксильных групп друг с другом. Молекулы воды при этом удаляются в газовую фазу. В силу этого процесс становится необратимым. Однако пространственная разобщенность гидроксильных групп при дегидратации приводит к тому, что на поверхности остаются анионный кислородный центр и катионный центр – силициниевый ион:  В ходе указанного процесса исчезают два кислотных Бренстедовских центра и появляются один основной и один кислотный Льисовских центры. Эти изменения в структуре поверхностного слоя фиксируются современными методами. Реакция каталитического крекинга инициируется возникшими при прокаливании алюмосиликатов силициниевыми ионами. Все процессы, происходящие при прокаливании алюмосиликатов, являются обратимыми. Поэтому, естественно, если прокаленный катализатор будет контактировать с парами воды, то все приведенные выше реакции будут протекать в обратном направлении. Это приведет к исчезновению катионных центров на поверхности алюмосиликатов. Соответственно, исчезает и их каталитическая активность. Взаимодействие силициниевых ионов со связями С-С или С-Н приводит к возникновению карбкатионов: 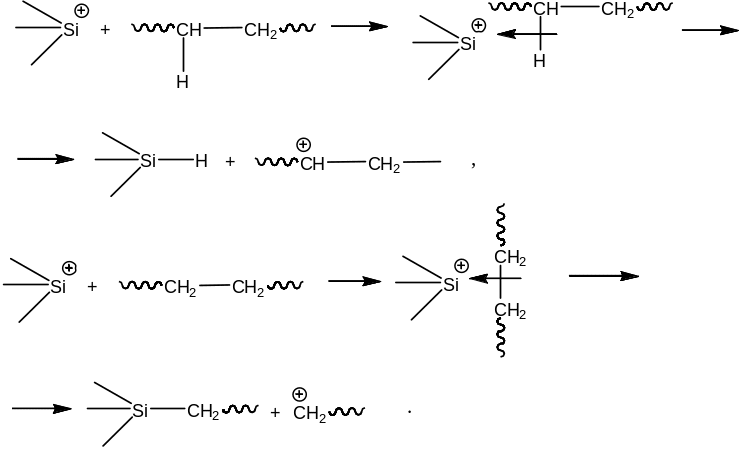 Дальнейший распад карбкатионов приводит к возникновению протонов, метильных катионов, которые являются продолжателями катионной цепи превращений. Однако все эти превращения продолжают протекать на поверхности алюмосиликата. Поэтому, когда частицы катализатора покрываются пленкой кокса, препятствующей адсорбции молекул углеводородов сырья на поверхности алюмосиликатов, то активность катализатора падает. Отсюда следует, что для поддержания высокой активности катализатора пленку кокса на его поверхности необходимо удалять. Это делается путем выжига этой пленки при высоких температурах в атмосфере воздуха. В ходе этой операции возникшие на поверхности катализатора алкилсилановые и силановые фрагменты окисляются до силанольных (силанолами называют соединения, содержащие фрагмент Si-OH). Структура алюмосиликата при этом полностью восстанавливается. Эволюция технологии каталитического крекинга. Приведенные выше данные указывали на то, что чем больше время контакта углеводородного сырья с катализатором, тем более вероятным становиться протекание побочных реакций. При этом происходит безвозвратная потеря первичных продуктов каталитического крекинга и возрастает образование кокса. Вся эволюция технологии каталитического крекинга является демонстрацией того, что с уменьшением времени контакта сырья с катализатором возрастал выход полезных продуктов. Первые установки каталитического крекинга (1930-1940 г.г.) имели реактора, в которых на полках находился неподвижный слой катализатора. Время контакта сырья с катализатором исчислялось часами. Эти установки давали только 18% полезных продуктов. Далее было осознано, что коль реакция крекинга проходит на поверхности катализатора, то целесообразно его на полках перемешивать. Появились установки с перемешивающимся слоем катализатора (1940-1954 г.г.). Время контакта при этом было сокращено до 10-15 минут. Проведенные совершенствования технологии привели к увеличению выхода полезных продуктов до 28%. Следующим этапом в развитии технологии каталитического крекинга явилось использование кипящего слоя катализатора (1954-1974 г.г.). Время контакта сырья с катализатором при этом было сокращено до 3-5 минут. Эти установки давали уже 42% полезных продуктов. Дальнейшим совершенствованием технологии каталитического крекинга явилось использование лифт-реакторов. Эти установки появились в 1974 г., и они частично эксплуатируются до настоящего времени. Время контакта на этих установках составляет менее 3 секунд. Выход полезных продуктов составляет 53 %. В лифт-реакторах, которые представлют собой обычную обогреваемую трубу, сырье и катализатор подают снизу, а продукты вместе с катализатором выводятся сверху. Цеолиты используют в виде микросферических частиц, средний диаметр которых составляет 60 мкм. Частицы размером до 40 и свыше 105 мкм не используют, так как слишком малые частицы уносятся из реактора, а крупные не обладают прочностью, достаточной для избежания истирания в потоке. За счет протекающих одновременно с крекингом процессов полимеризации образуется кокс и катализатор теряет активность. После реактора катализатор отделяется от смеси углеводородов на циклонах. Далее катализатор поступает в регенератор, в котором при 650-6700С в атмосфере воздуха проводят выжигание кокса, а выделяющееся при этом тепло используют для нагрева сырья. В новейших установках каталитического крекинга предусмотрена подача потоков катализатора и сырья во взаимно перпендикулярных направлениях. Эти установки получили название установок с ультракоротким временем контакта (УКВК). Оно доведено до миллисекунд. Эта технология позволяет конвертировать углеводородное сырье в полезные продукты с выходом 96%. Контрольные вопросы для проверки знаний по теме “Каталитический крекинг” 1. С позиций термодинамики обоснуйте температурные условия осуществления каталитического крекинга углеводородов. 2. В чем отличие карбониевых и карбениевых ионов? На примере этана покажите пути образования различных карбониевых ионов, и их пути распада до карбениевых ионов. 3. В чем заключается реакция β-распада карбениевых ионов? На примере первичного бутильного катиона (СН3СН2СН2СН2+) продемонстрируйте различные направления его β-распада. 4. н-Бутан в присутствии суперкислоты HF-TaF5 при 250С изомеризуется в изобутан, а н-пентан – в изопентан: CH3CH2CH2CH3 → (CH3)3CH , CH3CH2CH2CH2CH3 → (CH3)2CHCH2CH3 . Приведите схемы превращений для данных процессов. 5. Пониманию возможности вовлечения предельных углеводородов в кислотно-катализируемые реакции способствовал забавные случай, происшедший на рождественской вечеринке в лаборатории Нобелевского лауреата Д.Ола в Кливленде (штат Огайо, США). Его докторант И.Лукас случайно уронил в суперкислоту HSO3F-SbF5 кусок парафиновой свечи, который быстро исчез. Дайте объяснение этому явлению. 6. Среди продуктов каталитического крекинга н-гептана обнаруживаются пропилен, бутен-1, цис- и транс-бутены-2: СН3(СН2)5СН3 → СН3СН=СН2 + СН3СН2СН=СН2 + СН3СН=СНСН3. Приведите схемы превращений, объясняющих их образование. 7. При каталитическом крекинге линейных углеводородов в качестве побочных продуктов образуются нафтеновые углеводороды. Например, из пентана образуется циклопентан, из гексана – циклогексан, который далее превращается в бензол: 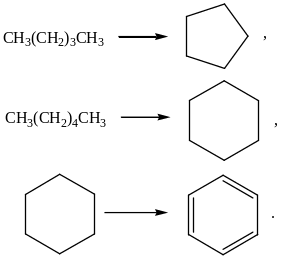 Приведите схемы превращений, объясняющих образование указанных продуктов. 8. Каталитический крекинг н-гексана частично сопровождается образованием гексатриена-1,3,5: СН3(СН2)4СН3 → СН2=СН−СН=СН−СН=СН2. Приведите схему превращений, объясняющую образование этого вещества. 9. При каталитическом крекинге алкилароматических углеводородов бензольное ядро не затрагивается. Боковые же цепи во всех случаях, кроме толуола, отщепляются с образованием олефинов. Энергия активации деалкилирования этилбензола составляет 209 кДж/моль: С6Н5СН2СН3 → С6Н6 + СН2=СН2, а изопропилбензола – 73 кДж/моль: С6Н5СН(СН3)2 → С6Н6 + СН3СН=СН2. Приведите стадии, через которые протекает деалкилирование алкилароматических соединений. Дайте объяснение наблюдаемым различиям в энергиях активации приведенных выше реакций. 10. Энтальпии образования различных карбениевых ионов, получаемых из н-гептана, составляют:
Чем обусловлено наблюдаемое различие в энтальпиях образования указанных ионов? Какой из атомов углерода в н-гептане предпочтительнее будет вовлекаться в превращение в условиях каталитического крекинга? Образования каких продуктов при этом можно ожидать? 11. Энергии активации каталитического крекинга углеводородов, протекающих по схеме: R-CH3 + H+ → R+ + CH4 , имеют следующие величины:
Чем обусловлен наблюдаемый ряд активности? 12. Дейтерометан СН3D в растворе суперкислоты HF-SbF5 превращается в смесь продуктов: CH3D → CH4 + CH3D + CH2D2 + CHD3 + CD4. Приведите схемы превращений, объясняющей появление указанных выше продуктов. 13. Конверсия изомерных гексанов в условиях каталитического крекинга в зависимости от строения их молекул меняется следующим образом:
Почему с увеличением степени разветвленности увеличивается конверсия алканов в процессе каталитического крекинга? 14. Какие процессы приводят к коксообразованию в процессах каталитического крекинга? Как влияет время контакта углеводородного сырья с катализатором на процесс коксообразования? 15. Коксообразование в процессе каталитического крекинга ускоряется, если исходное сырье содержит непредельные углеводороды. Это явление менее выражено тогда, когда сырье содержит только насыщенные углеводороды. Чем вызвано наблюдаемая зависимость скорости коксообразования от состава сырья? 16. Коксообразование в процессе каталитического крекинга резко возрастает при наличии в углеводородном сырье соединений тяжелых металлов. Чем обусловлено данное явление? 17. Двуокись кремния (SiO2) как катализатор не проявляет активности в процессе каталитического крекинга. Применение алюмосиликатов приводит к появлению каталитической активности. Чем вызвано различие в поведении двуокиси кремния и алюмосиликатов? Почему для появления каталитической активности у алюмосиликатов требуется их предварительный прогрев? 18. Будут ли проявлять в процессе каталитического крекинга каталитическую активность боросиликаты (продукты взаимодействия борной кислоты с гелем кремневой кислоты)? 19. Каталитическая активность алюмосиликатов в процессе каталитического крекинга зависит от соотношения Al/Si. С возрастанием содержания алюминия каталитическая активность алюмосиликатов сначала возрастает, а затем понижается. Чем обусловлено данное явление? 20. Активность алюмосиликатов как катализаторов в процессе каталитического крекинга подавляется, если углеводородное сырье содержит кислородсодержащие (вода, спирты, эфиры) и серосодержащие (меркаптаны, сульфиды) органические соединения. В чем причина этого явления? 21. История использования алюмосиликатов как катализаторов в процессах каталитического крекинга прошла этапы использования природных алюмосиликатов (глины), синтетических аморфных алюмосиликатов, синтетических кристаллических алюмосиликатов. Что явилось причиной указанной эволюции? 22. Каталитическая активность прогретых алюмосиликатов в процессе каталитического крекинга утрачивается, если катализатор контактировал со влажным воздухом. Чем это обусловлено? 23. Каталитическая активность цеолитов в процессе каталитического крекинга увеличивается при обработке их поверхности кислотами Льюиса (SnCl2, BF3, SbF5 и т.д.). При этом малые количества кислот Льюиса способствуют увеличению каталитической активности. Большие же количества приводят к полной дезактивации катализатора. Чем обусловлены наблюдаемые явления? Термический и каталитический крекинг. Риформинг – вторичная переработка нефти Бензина, получаемого при перегонке нефти, не хватает для покрытия всех нужд. В лучшем случае из нефти удается получить до 20% бензина, остальное – высококипящие продукты. В связи с этим перед химией стала задача найти способы получения бензина в большом количестве. Удобный путь был найден с помощью созданной А.М. Бутлеровым теории строения органических соединений. Высококипящие продукты разгонки нефти непригодны для употребления в качестве моторного топлива. Их высокая температура кипения обусловлена тем, что молекулы таких углеводородов представляют собой слишком длинные цепи. Если расщепить крупные молекулы, содержащие до 18 углеродных атомов, получаются низкокипящие продукты типа бензина. Этим путем пошел русский инженер В.Г.Шухов, который в 1891 г. разработал метод расщепления сложных углеводородов, названный впоследствии крекингом (что означает расщепление).  В.Г.Шухов (1853–1939) Сущность крекинга заключается в том, что при нагревании происходит расщепление крупных молекул углеводородов на более мелкие, в том числе на молекулы, входящие в состав бензина. Обычно расщепление происходит примерно в центре углеродной цепи по С—С-связи, например:  Однако разрыву могут подвергаться и другие С—С-связи. Поэтому при крекинге образуется сложная смесь жидких алканов и алкенов. Получившиеся вещества частично могут разлагаться далее, например: 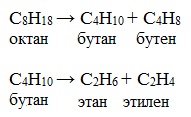 Такой процесс, осуществляемый при температуре около 470°С – 550°С и небольшом давлении, называется термическим крекингом. Этому процессу обычно подвергаются высококипящие нефтяные фракции, например, мазут. Процесс протекает медленно, при этом образуются углеводороды с неразветвлённой цепью атомов углерода. Бензин, получаемый термическим крекингом, невысокого качества, не стоек при хранении, он легко окисляется, что обусловлено наличием в нём непредельных углеводородов. Однако, детонационная стойкость (взрывоустойчивость, характеризующаяся октановым числом) такого бензина выше, чем у бензина прямой перегонки из-за большого содержания непредельных углеводородов. При использовании, к бензину необходимо добавлять антиокислители, чтобы защитить двигатель. Коренным усовершенствованием крекинга явилось внедрение в практику процесса каталитического крекинга. Этот процесс был впервые осуществлен в 1918 г. Н.Д.Зелинским.  Н.Д.Зелинский (1861–1953) Каталитический крекинг позволил получать в крупных масштабах авиационный бензин. Его проводят в присутствии катализатора (алюмосиликатов: смеси оксида алюминия и оксида кремния) при температуре 450 — 500°С и атмосферном давлении. Обычно каталитическому крекингу подвергают дизельную фракцию. При каталитическом крекинге, который осуществляется с большой скоростью, получается бензин более высокого качества, чем при термическом крекинге. Это связано с тем, что наряду с реакциями расщепления происходят реакции изомеризации алканов нормального строения. Кроме того, образуется небольшой процент ароматических углеводородов, улучшающих качество бензина. Бензин каталитического крекинга более устойчив при хранении, так как в его состав входит значительно меньше непредельных углеводородов по сравнению с бензином термического крекинга, обладает ещё большей детонационной стойкостью, чем бензин термического крекинга. Сравнение термического и каталитического крекинга Учебный фильм «Каталитический крекинг нефти» Таким образом, высокое качество бензина, получаемого каталитическим крекингом, обеспечивается наличием в его составе разветвленного строения углеводородов и ароматических углеводородов. Бензин: состав и октановое число. Детонация 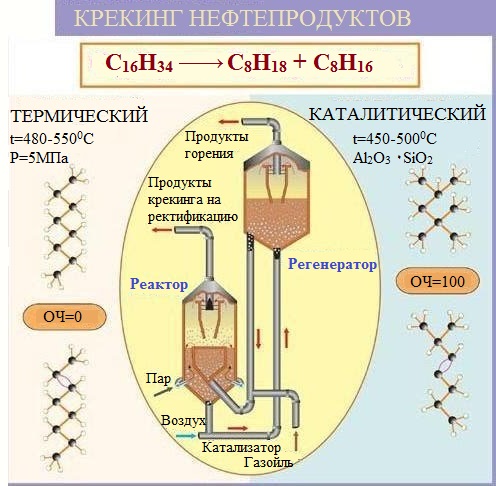 Основным способом переработки нефтяных фракций являются различные виды крекинга. Впервые (1871–1878) крекинг нефти был осуществлен в лабораторном и полупромышленном масштабе сотрудником Петербургского технологического института А.А.Летним. Первый патент на установку для крекинга заявлен Шуховым в 1891 г. В промышленности крекинг получил распространение с 1920-х гг. Крекинг – это термическое разложение углеводородов и других составных частей нефти. Чем выше температура, тем больше скорость крекинга и больше выход газов и ароматических углеводородов. Крекинг нефтяных фракций кроме жидких продуктов дает первостепенно важное сырье – газы, содержащие непредельные углеводороды (олефины). Различают следующие основные виды крекинга: жидкофазный (20–60 атм, 430–550°С), дает непредельный и насыщенный бензины, выход бензина порядка 50%, газов 10%; парофазный (обычное или пониженное давление, 600°С), дает непредельно-ароматический бензин, выход меньше, чем при жидкофазном крекинге, образуется большое количество газов; пиролиз нефти – разложение органических веществ без доступа воздуха при высокой температуре (обычное или пониженное давление, 650–700°С), дает смесь ароматических углеводородов (пиробензол), выход порядка 15%, более половины сырья превращается в газы; деструктивное гидрирование (давление водорода 200–250 атм, 300–400°С в присутствии катализаторов – железа, никеля, вольфрама и др.), дает предельный бензин с выходом до 90%; каталитический крекинг (300–500°С в присутствии катализаторов – AlCl3, алюмосиликатов, МоS3, Сr2О3 и др.), дает газообразные продукты и высокосортный бензин с преобладанием ароматических и предельных углеводородов изостроения. В технике большую роль играет так называемый каталитический риформинг – превращение низкосортных бензинов в высокосортные высокооктановые бензины или ароматические углеводороды. 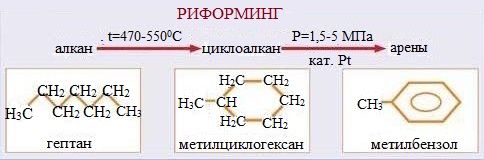 Основными реакциями при крекинге являются реакции расщепления углеводородных цепей, изомеризации и циклизации. Огромную роль в этих процессах играют свободные углеводородные радикал |