лекции 1-10 по химии поверхности (1). Лекция 1 Общие представления о поверхности твердых тел
 Скачать 1.48 Mb. Скачать 1.48 Mb.
|

25 мэВ). Лекция 4 Адсорбция на поверхности твердых тел Как уже отмечалось выше одним из путей восстановления насыщенного характера связей поверхностных атомов является адсорбция атомов или молекул из внешней среды. Рассмотрим этот процесс более подробно и приведем некоторые основные определения. Твердое тело, на котором происходит поглощение газов и паров, называется адсорбентом, а адсорбированное вещество – адсорбатом. Процесс, обратный адсорбции, называется десорбцией. Величину адсорбции, то есть количество адсорбированного газа (или пара), выражают в разных единицах, но наиболее часто в молях адсорбированного вещества на 1 г адсорбента. Понятно, что величина адсорбции данного вещества тем выше, чем более доступна для этого вещества поверхность адсорбента. Поэтому в качестве характеристики твердых тел приводят величину удельной поверхности S (площадь поверхности 1 г адсорбента). Физическая и химическая адсорбция. Традиционно считают, что молекулы могут адсорбироваться на поверхности двумя способами. В случае физической адсорбции взаимодействие между поверхностью и адсорбированной молекулой обусловлено межмолекулярным взаимодействием, которое не приводит к разрыву или образованию новых химических связей. При этом молекула сохраняет свою индивидуальность, хотя, вероятно, может быть растянута или изогнута из-за близости поверхности. Такие взаимодействия часто называют Ван-дер-ваальсовыми. К особенностям межмолекулярных взаимодействий при адсорбции, отличающих их от взаимодействий между молекулами в газах, относится весьма тесное сближение молекул адсорбата с атомами, ионами или функциональными группами, образующими поверхность адсорбента, а при относительно больших величинах адсорбции даже и между собой. Поэтому явление адсорбции часто имеет много общего с конденсацией пара и молекулярной ассоциацией в жидкостях. Кроме того, адсорбированная молекула взаимодействует не с одним центром на поверхности адсорбента, а со многими соседними центрами. Глубина потенциальной ямы физической адсорбции невелика и меньше, чем энергия химической связи ( |
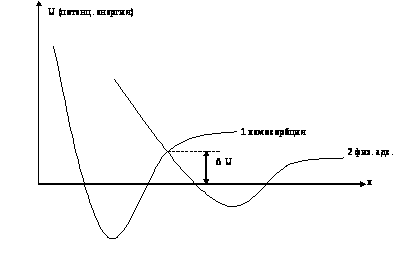 |
Рис. 4.1. Соотношение энергии химической (1) и
физической (2) адсорбции
Экспериментальное определение величины адсорбции. При экспериментальном определении величины адсорбции вначале необходимо очистить поверхность от ранее адсорбированных веществ. Для этого исследуемый материал предварительно прогревают в вакууме при давлении ниже 10–3 Па (в случае статических методов) или в токе инертного газа (если применяются газохроматографические методы). Далее, если твердому телу дать возможность войти в контакт с газом или паром вещества, количество которого до адсорбции нам известно, то поверхность тела начинает адсорбировать газ (пар) и его давление уменьшается. Спустя некоторое время давление p становится равновесным, то есть постоянным при заданной температуре опыта. Основываясь на законах идеальных газов, если известен свободный объем сосуда, в котором находится адсорбент, можно по снижению давления рассчитать количество адсорбированного вещества. Такие статические методы исследования адсорбции называют объемными. В случае весовых методов величину адсорбции при данном p определяют просто по увеличению массы адсорбента, используя высокочувствительные весы.
Для данного адсорбата и адсорбента равновесная величина адсорбции а газа или пара определяется температурой Т и давлением p. При исследовании адсорбции одну из этих величин обычно поддерживают постоянной. Чаще всего в результате непосредственных измерений получают зависимость а от р при постоянной температуре, которая носит название изотермы адсорбции. Формы изотермы адсорбции чрезвычайно разнообразны. Измеряя изотермы адсорбции при равных Т, можно получить зависимость р от Т для данной величины адсорбции – изостеру адсорбции.
Развитие теории адсорбционных сил еще не достигло такой стадии, когда по известным физико-химическим свойствам газа и твердого тела можно было бы рассчитать изотерму адсорбции, не проводя экспериментальных исследований. Поэтому попыткам описать экспериментальные изотермы с помощью различных теоретических уравнений, которым соответствуют определенные модели адсорбции, посвящено огромное количество работ. Если теоретическое уравнение изотермы адсорбции хорошо воспроизводит экспериментальные данные, то можно рассчитать неизвестные величины адсорбции при разных условиях (р и Т) и определить различные геометрические параметры твердых тел.
Рассмотрим наиболее распространенные теоретические уравнения изотерм адсорбции.
Модель Ленгмюра. Основные положения, лежащие в основе вывода изотермы адсорбции согласно модели Ленгмюра, предложенной в 1916 г., заключаются в следующем: 1) поверхность адсорбента однородна, то есть теплота адсорбции на разных участках поверхности одинакова; 2) теплота адсорбции не зависит от присутствия других адсорбированных молекул, следовательно, можно пренебречь взаимодействием адсорбированных молекул между собой; 3) молекулы не могут адсорбироваться на молекулах первого слоя и максимальная адсорбция, которую обозначают как аm, наблюдается при полной упаковке адсорбированных молекул на поверхности в слое толщиной в одну молекулу (рис. 4.2).
Рис. 4.2. Модель адсорбции Лэнгмюра
Предполагая, что адсорбированные молекулы находятся в динамическом равновесии с молекулами в газовой фазе, процесс адсорбции можно описать с помощью схемы: «Молекулы в газе + Свободная поверхность = Адсорбированные молекулы». Такая модель приводит к известному уравнению изотермы Ленгмюра:
где а- величина адсорбции, Кр – константа адсорбционного равновесия, αm – общее число мест поверхности.
В области малых давлений К<<1 и уравнение преобразуется в уравнение изотермы Генри:
Величина аm позволяет вычислять удельную пористость адсорбентов. Если известна площадка ω, которую занимает молекула в монослое, то можно рассчитать удельную поверхность S адсорбента по уравнению:
где Na – постоянная Авогадро.
В настоящее время для обработки экспериментальных изотерм с целью определения величины S предпочитают использовать более надежное уравнение БЭТ.
Модель БЭТ. Знаменитая теория полимолекулярной адсорбции Брунауэра, Эммета и Теллера (1936 г.), получившая название теории БЭТ по первым буквам фамилий ученых, основана на модели адсорбционного процесса, предложенной Ленгмюром. Однако в модели БЭТ учтена возможность адсорбции как на первом, так и на втором, третьем и т. д. слое уже адсорбированных молекул и, следовательно, возможность полимолекулярной адсорбции. При построении модели полагают, что энергия адсорбции равна теплоте конденсации паров для слоев выше первого. Такой подход позволяет вывести уравнение адсорбции БЭТ:
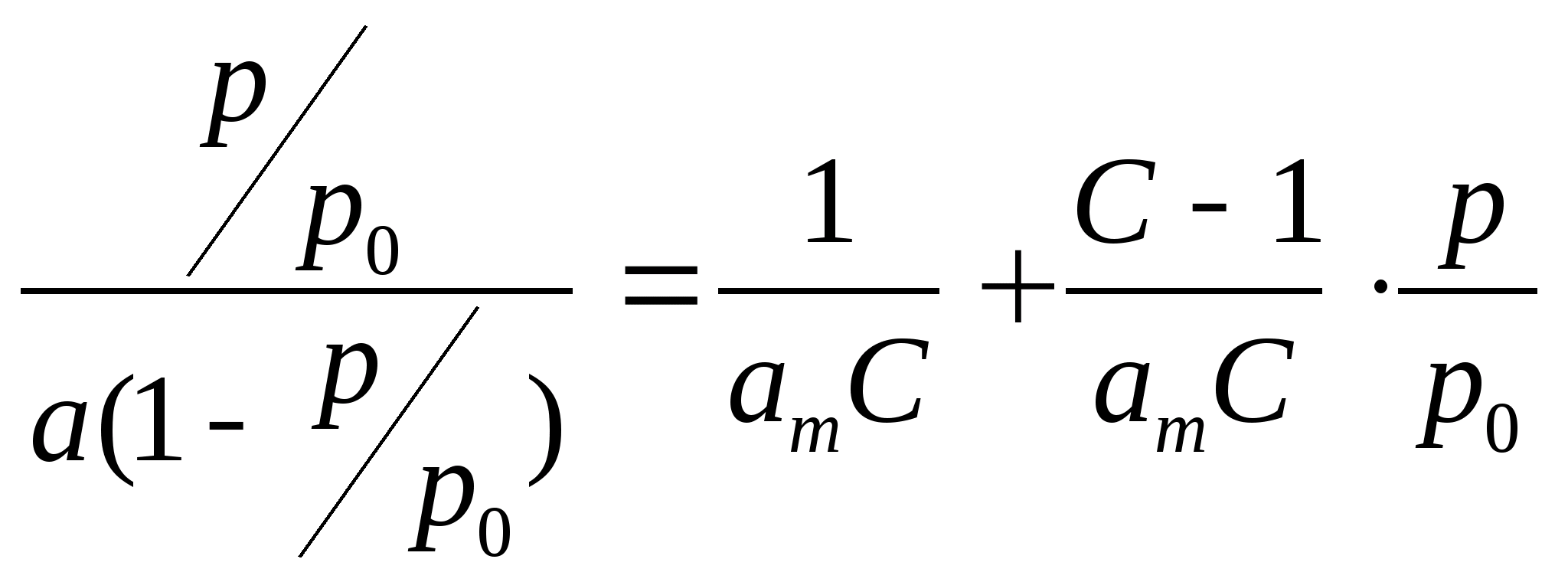 (4.4)
(4.4)где С – постоянная данной адсорбционной системы, связанная с теплотой и энтропией адсорбции, р0 – давление насыщенных паров адсорбата.
При С>>1 и небольших значениях р уравнение БЭТ переходит в уравнение Ленгмюра. В качестве адсорбатов для определения удельной поверхности твердых тел по методу БЭТ из экспериментальных изотерм, измеренных при температуре кипения жидкого азота (77 К), наиболее подходящими являются азот или криптон с «посадочной» площадкой молекулы 0,16 и 0,2 нм2 соответственно.
Модель БЭТ неоднократно критиковали, поскольку трудно представить себе абсолютно однородные поверхности. Кроме того, реальные экспериментальные данные свидетельствуют о боковых взаимодействиях адсорбированных молекул между собой. Некоторые исследователь пытались усовершенствовать теорию БЭТ или получить другие модельные изотермы, однако и по сей день метод БЭТ является основным для определения удельной поверхности твердых тел, в том числе адсорбентов и катализаторов. Следует отметить, что адсорбционный метод является практически единственным способом определения величины поверхности непористых порошков и лучшим методом расчета внутренней поверхности пористых твердых тел, если, конечно, поры доступны по своим размерам для адсорбции исследуемых молекул.
Особенности взаимодействия адсорбатов с поверхностью твердых тел. При рассмотрении взаимодействия адсорбатов с поверхностью твердых тел необходимо учитывать силы притяжения и силы отталкивания, быстро увеличивающиеся при уменьшении расстояния. Силы притяжения определяются взаимодействием следующих типов: а) дисперсионным, связанным с согласованным движением электронов в сближающихся молекулах; б) ориентационным, наблюдающимся в случае адсорбции полярных молекул на заряженных поверхностях; в) индукционным, связанным с появлением наведенных дипольных моментов в адсорбированных молекулах или адсорбенте. В случае хемосорбции наблюдается также перенос электронов между твердым телом и адсорбатом и/или совместное обладание электронами.
Хемосорбция на поверхности металлов. Наиболее простую из возможных трактовок переноса заряда и связи с поверхностью металла дает модель резонансного уровня. Адсорбат в ней рассматривается как потенциальная яма, содержащая единственное связанное состояние с заданной энергией. На большом расстоянии адсорбат и адсорбент сохраняют свою индивидуальность. При сближении происходит перекрывание волновых функций адсорбированного атома и подложки, появляется собственное состояние системы, узкий атомный уровень превращается в более широкий резонансный, который «просачивается» (по туннельному механизму) в металл. Заселенность резонансного уровня адсорбата и характер поверхностной связи зависят от относительного расположения энергии резонанса и уровня Ферми в металле. Если резонансный уровень адсорбата расположен ниже уровня Ферми, то образуется ионная связь и происходит перенос заряда (рис. 4.3).
Хемосорбция кислорода на металлах фактически приводит к образованию новой фазы, т.е. оксидной пленки. В то же время продукты хемосорбции О2 отличаются от соответствующих оксидов. Например, WO3 восстанавливается водородом, а соединение, образующееся при адсорбции кислорода на вольфраме, не реагирует с водородом даже при 1600 0С.

Рис. 4.3. Зонная модель адсорбции кислорода на поверхности металла
Термодинамические расчеты показывают, что все металлы должны самопроизвольно окисляться при низких давлениях кислорода (10–6 мм рт. ст.) при комнатной температуре в соответствии с уравнением:
Ме (тв) + ½ О2 (г) → МехО (тв); ΔG < 0. (4.5)
На большинстве чистых металлов хемосорбированный слой кислорода образуется даже при температурах ниже комнатной.
К недостаткам модели резонансного уровня относится то, что она пренебрегает деталями электронной структуры молекулы адсорбата и не применима к переходным металлам, имеющим связанные электроны.
Наибольшей активностью при хемосорбции обладают переходные металлы, что связано с участие d-орбиталей в образовании адсорбционных связей. Для лучшего понимания механизма связывания адсорбатов на d-орбиталях желательно определить, какие из лепестков d-орбиталей на поверхности металла доступны для образования связей. Это определяется кристаллическая структурой и изучаемой гранью кристалла и допущением о постоянном положении атомов на поверхности. Пользуясь этим методом, можно классифицировать металлические катализаторы5. Если принять, что обычно внешняя поверхность представляет собой в случае кубической и гексагональной плотноупакованной структур грань (111), а в случае объемноцентрированного кубического кристалла – грань (110) то получим следующие результаты: класс I – Mo, W (вакантная d-орбиталь, перпендикулярная поверхности); класс II – Rh, Ir, Ru, Os, Tc, Re (вакантная d-орбиталь под углом 35–45 0 к поверхности); класс III – Fe, Co, Ni, Pd, Pt (частично занятые d-орбитали под углом 30–50 0 к поверхности); класс IV – Zn, Ga, Cd, In, Sn, Pb (ассиметрия d-оболочки).
Высокую каталитическую активность элементов класса III можно приписать тому, что поверхность этих металлов при доступности частично занятых d-орбиталей способна образовывать связи с олефинами (настолько прочные, что этилен, например, разрушается) путем перекрывания молекулярных π-орбиталей олефина и d-орбиталей металла:
π-электроны → d-орбиталь
Другой вариант связывания возникает при адсорбции оксида углерода (II) СО на металлах. ИК-спектроскопические исследования показали, что СО связан с поверхностью металла σ-связью, при этом имеет место обратный переход d-электронов на свободную π-молекулярную орбиталь молекулы СО:
π-орбиталь ← d- электроны
Хемосорбция на полупроводниках и диэлектриках может приводить к насыщению оборванных связей путем образования локальной химической связи на поверхности, если имеется локализованная орбиталь или происходит локализация захваченного электрона (рис. 4.4).
| O O O ║ ║ ║ ─ Ge ─ Ge ─ Ge ─ │ │ │ ─ Ge ─ Ge ─ Ge | O2- ─ Ge ─ Ge ─ Ge ─ │ │ │ ─ Ge ─ Р+ ─ Ge ─ │ │ │ ─ Ge ─ Ge ─ Ge ─ |
| Рис. 4.4. Схема хемосорбции кислорода на поверхности германия с образованием ковалентной связи | Рис.4.5. Схема ионосорбции на поверхности полупроводника (противоположный заряд на атоме фосфора) |
К этому же типу относится адсорбция на поверхности ионных твердых тел, имеющих кислотные или основные центры. Образуется кислотно-основная ковалентная связь с соответствующими основанием или кислотой из газовой фазы. Хемосорбции данного типа отвечает пренебрежимо малая энергия активации, так как адсорбат обычно хемосорбируется в виде молекулы и связанная с ее диссоциацией энергия активации отсутствует; кроме того, взаимодействие между молекулами адсорбата мало, поскольку каждая молекула взаимодействует только со своим активным центром, а в случае очень сильных твердых кислот или оснований близко расположенных активных центров нет.
На поверхности полупроводников возможен и процесс ионосорбции, связанный с ионизацией адсорбированного атома или молекулы электроном из зоны проводимости (или дыркой из валентной зоны) (рис. 4.5).
При этом противоположно заряженные ионы могут находиться за сотни Å от места расположения адсорбированных ионов, а адсорбированные частицы рассматриваются как поверхностные состояния. На поверхности полупроводников возникает двойной электрический слой.
Лекция 5
Поверхностные центры кислотного и основного типа
Природа поверхностных центров. На поверхности твердого тела возможно существование двух видов кислотных и основных центров – центров Льюиса и центров Бренстеда. Наличие кислотных и основных центров (а также воды) на поверхности будет определять химическую активность твердого тела как катализатора, в процессах коррозии, в электрохимических процессах, включая процессы переноса электрона на поверхности, в фотоэлектро- и фотохимических процессах и т.д.
Аналогично молекулярной кислоте Льюиса кислотный поверхностный центр Льюиса (Ls+) – это такой центр на поверхности, который имеет свободную орбиталь с высокой энергией сродства к паре электронов, т.е. является акцептором электронной пары. Поэтому, когда адсорбируемая основная молекула (например, NH3) делится с центром своей электронной парой, происходит значительное уменьшение энергии.
На поверхности могут возникать также основные центры Льюиса (L-OH). Эти центры имеют электронные пары на орбиталях с высокой энергией, т.е. являются донорами электронной пары. При распределении электронной пары между поверхностным центром и адсорбируемым акцептором происходит значительное уменьшение энергии.
Кислотные центры Бренстеда (HS+) имеют тенденцию отдавать протон, а основные центры Бренстеда выступают в качестве акцепторов протона.
В присутствии воды кислотные центры Льюиса могут переходить в кислотные центры Бренстеда и наоборот:
L+ + H2O
Вступая в качестве катиона в реакцию на поверхности центр Льюиса (L+) делит электронную пару с ионом гидроксила ОН–, а остающийся свободным и адсорбируемый на поверхности протон HS+ легко может вступать в химические реакции. Таким образом, активность по Льюису может переходить в активность по Бренстеду.
Необходимо отметить, что твердое тело, например, оксид должен обладать двумя типами центров даже в случае однородной поверхности. На поверхности оксида незанятые орбитали катионов будут действовать как акцепторные поверхностные состояния или кислотные центры Льюиса, а орбитали ионов кислорода будут представлять собой донорные поверхностные состояния или основные центры Льюиса.
В результате адсорбции воды на поверхности оксида также будут представлены два класса центров, но теперь это центры по Бренстеду. Ион ОН–, прочно соединяясь с катионом, становится основным центром по Бренстеду, а остающийся протон на решеточном ионе кислорода становится кислотным центром по Бренстеду. Соединенные с катионами группы ОН– могут проявлять и слабые кислотные свойства, при этом также может быть отдан протон. Поэтому центры такого типа могут проявлять себя и как слабокислотные центры в присутствии достаточно сильного основания. Следует ожидать, что кислотность находящихся на катионах групп ОН– будет всегда слабее, чем кислотность ионов Н+, адсорбированных на решеточных ионах кислорода.
При адсорбции воды на поверхности ионных твердых тел, наряду со связыванием на центрах Льюиса, может происходить поляризация молекул Н2О в электрическом поле поверхности твердого тела (например, в кристаллическом поле иона переходного металла) с последующим связыванием. Возможно также образование «водородной связи», когда один из ионов водорода осуществляет связывание между ионом кислорода воды и анионом твердого тела. Перечисленные типы связывания представляют собой просто различные вариации на одну и ту же тему, причем редко какой-либо из них встречается в чистом виде.
Методы исследования кислотности поверхностных центров. Поскольку наличие адсорбированной воды, а также различных типов центров на поверхности твердых тел определяет их электрохимическую и химическую активность, то целесообразно рассмотреть методы экспериментального исследования кислотности поверхности. Большинство методов измерений не позволяют отличить активность Льюиса от активности Бренстеда. Сила кислотного центра определяется способностью превратить адсорбируемую молекулу кислоты в сопряженное ей основание. Поэтому в случае Льюиса сила кислотного или основного центра тесно связана с величиной сродства данных центров к электрону. При таком определении сила кислотного центра не определяется однозначно его сродством к электрону, а зависит также от геометрии центра и пространственной ориентации свободных орбиталей. Одновременно кислотность центра зависит от тех же характеристик адсорбируемой молекулы основания. Поэтому, если ряд твердых тел расположить в определенном порядке по степени кислотности, полученной в результате измерений с одним каким-нибудь молекулярным основанием, то при другом молекулярном основании этот порядок может не сохраниться. Поэтому измерения кислотности или основности поверхностных центров должны рассматриваться как полуколичественные.
Например, установлено, что в реакции гидратации пропилена кислотность поверхности катализаторов понижается в следующем ряду:
Fe2(SO4)3 Al2(SO4)3 Cr2(SO4)3 CuSO4 ZnSO4 CoSO4 CdSO4 NiSO4 MnSO4 K2SO4 Na2SO4.
При этом для предварительной оценки кислотности поверхности можно воспользоваться формулой:
a (ra/rc) / Z2, (5.2)
где а – кислотная функция, ra – радиус аниона, rc – радиус катиона, Z – формальный заряд катиона.
Наиболее известным и экспериментально наиболее простым методом исследования поверхности является метод цветовых индикаторов. Рассмотрим его более подробно.
Сила кислоты Бренстеда на поверхности твердого тела характеризуется способностью этого центра сдвигать вправо реакцию:
Нs+ + В
где В – молекула основания, ВНs+ - сопряженная ей кислотная форма.
Константа равновесия для рассматриваемой системы может быть определена по уравнению:
Если обозначим кислотность поверхностного центра по Бренстеду Н0 и определим, что Н0 = –lg[Hs+], то отношение основной и кислотной форм адсорбированного основания можно выразить через кислотность поверхностных центров Бренстеда:
Когда [BH+] = [B] , H0 – pK = 0, H0 = pK.
Аналогично определяется кислотность центра Льюиса Н0 = – lg аА, где аА – активность акцептора электронной пары.
Выражение (5.5) можно записать в форме:
где В – основание, а через АВs обозначена такая форма основания, при которой его электронная пара обобществлена.
Смысл приведенных уравнений становится ясным при использовании кислотно-основных индикаторов, которые меняют цвет в зависимости от отношения концентраций в явных частях уравнений так, что по цвету адсорбированного индикатора можно сразу определить больше Н0, чем рК индикатора или меньше. Для того, чтобы измерить кислотность поверхности, индикаторы в основной форме с различным значением рК (например, фенилазонафтиламин с рК 4,0 или метилрот с рК 4,8) диспергируют в инертном растворителе, а исследуемое твердое тело помещают по очереди в каждый индикатор, чтобы определить будет ли индикатор адсорбирован в кислотной форме. С использованием данного метода было установлено, что, например, на поверхности TiO2 присутствует от трех до семи типов кислотных центров.
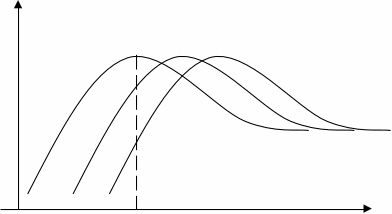
В случае адсорбции газообразных кислот или оснований достаточно прямым способом определения энергии связи частиц, адсорбированных на поверхности, является метод термостимулированной десорбции. Если повышать температуру твердого тела с постоянной скоростью, то первыми с поверхности будут десорбироваться частицы с самой низкой энергией активации десорбции, а затем – со все более высокой (рис. 5.1). Изменяя скорость нагревания так, чтобы получить пики при различных Тр, можно определить энергию активации адсорбции.
Разработана модель, в которой температура, соответствующая пику скорости десорбции (Тр), связана со скоростью нагревания β (град/с) уравнением:
где Е– энергия активации адсорбции, А – константа.
Рассматриваемый метод количественно применим только в тех случаях, когда адсорбат характеризуется одним или двумя моноэнергетическими адсорбционными центрами. Если имеется спектр энергий активации десорбции, то анализ результатов может стать очень сложным.
Наиболее селективным методом изучения природы кислотности поверхности в настоящее время является колебательная спектроскопия. Использование этого метода позволяет определить тип и свойства поверхностных центров в результате исследования ИК-спектров взаимодействующих с ними электронодонорных молекул. В качестве адсорбата в этом методе используют молекулы, которые способны специфически взаимодействовать с кислотными поверхностными центрами, при этом их спектр должен быть чувствителен к типу цента. Кроме того молекулы не должны участвовать в поверхностных реакциях, приводящих к изменению свойств поверхности. Чаще всего в качестве таких зондовых молекул используют аммиак, пиридин, оксид углерода (II),оксид азота (II),водород и др. молекулы. Например, при изучении адсорбции аммиака на поверхности диоксида титана установлено, что адсорбция на кислотных центрах Льюиса приводит к образованию ионов NH4+, характеризующихся полосой поглощения при 1450–1480 см–1, а в случае адсорбции на кислотных центрах Льюиса происходит координационное связывание аммиака, описываемое полосами поглощения при 1160 и 3420 см–1. Аналогичным образом в случае диоксида титана адсорбция пиридина на поверхностных центрах Бренстеда приводит к появлению в ИК-спектре полосы поглощения при 1540 см–1.
V(p)
 T
TTp
Рис. 5.1.. Зависимость объема (давления) от температуры
при десорбции для разных скоростей нагрева образца
Изменение спектра адсорбированных молекул позволяет характеризовать относительную силу кислотности электроноакцепторных центров оксидов. Например, в результате анализа положения полосы колебания координированного акцепторными центрами молекул аммиака было установлено, что кислотность Льюиса поверхности оксидов изменяется следующим образом:
Al2O3 > Ga2O3 > TiO2, Cr2O3, ZnO > ZrO2 > MgO > Ni2O3 > NiO, CuO.
Все рассмотренные методы исследования поверхностных центров позволяют получить информацию о наличии и типах центров, но не их концентрации на поверхности. В настоящее время наиболее остро стоит задача разработки стандартных методик определения природы и силы кислотных центров поверхности.
Лекция 6
Химическое модифицирование поверхности твердых тел
Общие представления о модифицировании. При сорбции в той или иной степени происходит изменение свойств как сорбента, так и сорбата. Система «вещество на носителе» зачастую представляет собой новый материал с рядом свойств, которыми не обладали ни носитель, ни сорбированное соединение. Под химическим модифицированием поверхности понимают химические превращения поверхностных функциональных групп, не затрагивающие остов носителя. Такой подход позволяет разграничить химическое модифицирование и гетерогенные химические реакции, которые распространяются вглубь носителя или приводят к наращиванию остова6.
В качестве носителей для закрепления различных соединений используют как органические (целлюлоза, декстран, полиакриламид, полиаминостирол и др. синтетические полимеры), так и минеральные (металлы, уголь, оксиды, карбонат кальция, стекло, глина и др.) подложки, содержащие на поверхности структурные функциональные группы. В случае полимерных подложек – это функциональные группы основной или боковой цепи полимера, в случае металлов – атомы металла, локализованные в поверхностном слое. В большинстве случаев в качестве носителей при химическом модифицировании используют неорганические оксиды, в частности кремнезем, на поверхности которых имеются гидроксильные группы, связанные с атомами остова носителя. К преимуществам неорганических носителей относятся: химическая стойкость, механическая прочность, устойчивость к набуханию в различных растворителях, термическая стабильность, радиационная устойчивость, высокая скорость массообмена. Носители могут различаться не только по своей химической природе, но и по морфологии (кристаллические или аморфные), по структуре (монокристаллические, высокодисперсные, пористые и т.п.).
В качестве модификаторов, реагирующих с поверхностными функциональными группами, могут использоваться кремнийорганические соединения (например, для повышения гидрофобности и пластической прочности поверхности), органические красители (для повышения светочувствительности в видимой области спектра), ионы и мелкие частицы металлов (для изменения электро- и фотоэлектрохимических свойств, повышения каталитической активности), комплексы с переносом заряда, ферменты, полимеры и др.
Необходимо отметить, что до недавнего времени химическое модифицирование проводилось с целью изменения в нужную сторону свойств поверхности носителя; оно ставило своей задачей достижение заданных физико-химических, механических или иных свойств твердого тела. При этом природа модификатора не играла первостепенной роли. Иначе обстоит дело, когда определяющую роль в системе «вещество на носителе» играет нанесенный компонент. Так, при иммобилизации ферментов, синтезе гетерогенных металлокомплексных катализаторов, закреплении на поверхности носителя комплексантов, существенно важна природа нанесенного соединения, тогда как природа носителя уходит на второй план. Отсюда возникла необходимость в новом термине, обозначающем фиксацию на поверхности носителя химических соединений, находящихся в газообразной или жидкой фазе, с целью получения материала, свойства которого преимущественно определяются природой фиксируемого соединения. В области химии и биологии употребляют много терминов: «иммобилизация» (в основном применительно к ферментам), «закрепление» и «гетерогенизация» в катализе, «прививка» в химии высокомолекулярных соединений. В целом же речь идет о химическом модифицировании поверхности твердого тела.
Способы химического модифицирования. Фиксация активного компонента на поверхности носителя может осуществляться различными способами: 1) обратимая адсорбция реагента на поверхности; 2) включение реагента в матрицу геля, пасты или полимера и, как частный случай, микрокапсулирование; 3) закрепление реагента на поверхности посредством образования химических связей. Рассмотрим эти способы несколько подробнее.
Обратимая адсорбция реагентов на поверхности твердого тела. Этот способ является наиболее старым и широко используется, например, при адсорбции красителей на поверхности полупроводников с целью изменения их спектральных характеристик, или при адсорбции ферментов на активированном угле или геле гидроксида алюминия.7 При модифицировании носителей путем адсорбции на их поверхности полимерных материалов отсутствует непосредственная химическая связь модификатора с носителем, однако в большинстве случаев полимерный чехол не может быть снят с частицы без ее разрушения, т.к. они представляют собой единое целое. Если при этом используются пористые носители, например, кремнезем, то по мере увеличения толщины выстилающей поверхность кремнезема полимерной пленки происходит постепенное заполнение пор кремнезема, и он фактически превращается в полимер, наполненный кремнеземом. В процессе адсорбции биополимеров на поверхности носителей можно выделить три стадии: первая стадия – гидрофобное взаимодействие, затем – частичная денатурация и разворачивание белковой глобулы, затем – необратимая адсорбция.
Независимо от природы модификатора, необратимая хемосорбция, идущая при непрерывном притоке реагента и удалении летучих продуктов, с течением определенного времени приводит к 100 % заселению поверхности сорбента (подложки) соответствующими функциональными группами. Продуктом такой исчерпывающей хемосорбции является монослой сорбированных структурных единиц, который при заданных условиях имеет постоянный состав.
Частным случаем рассматриваемого способа модифицированияявляются пленки Лэнгмюра–Бюджетт (названы в честь И. Лэнгмюра и его ассистентки К. Блоджетт, разработавших в 1930 г. лабораторные приемы их создания). Эти пленки представляют собой мономолекулярные структуры толщиной 2–3 нм, упорядоченно ориентированные на поверхности. Как правило, это молекулы с гидрофильной «головкой» и длинным углеводородным (жирным) «хвостом» (рис. 6.1). В настоящее время разработана технология переноса их на подложку, надстройки и взаимной ориентации (рис. 6.2).
Способ модифицирования путем включения реагента в матрицу пасты, геля или полимера также используется достаточно давно. Примером такой системы может быть угольно-пастовый электрод – паста, полученная смешением угольного порошка (графит) с маслом, в которую вставлена медная проволока. Добавление модификаторов используется для создания биосенсоров, например, при включении ферроцена в угольно-пастовый электрод получают глюкозный биосенсор.
Рис.6.1. Структура пленок Лэнгмюра–Блоджетт
Модифицировать поверхность можно, осаждая на нее тонкие слои оксидов металлов с использованием процессов гидролиза (так называемая золь-гель технология). Подробнее эти процессы рассматриваются в разделе «Химия тонких пленок». Использование золь-гель процесса позволяет осуществлять закрепление модификатора в трехмерной сетке геля путем инкапсулирования. При этом молекулы модификатора, чаще всего органического вещества, включаясь в сетку геля, не теряют своей химической индивидуальности. Рассматриваемые системы служат основой для получения ормосилов (Ormosil – organically modified silica). Возможно также включение модификаторов в сетку органических полимерных материалов. Вариантами метода являются микрокапсулирование и модифицирование с использованием мембранных технологий8.
Рис. 6.2. Молекулярные структуры на основе пленок
Лэнгмюра-Блоджетт: ()
Во всех рассматриваемых случаях модифицирование путем включения реагента в массу носителя обеспечивает более высокую концентрацию модификатора и улучшение эксплуатационных свойств материала.
Лекция 7
ЗАКРЕПЛЕНИЕ РЕАГЕНТОВ НА ПОВЕРХНОСТИ ПОСРЕДСТВОМ ОБРАЗОВАНИЯ ХИМИЧЕСКИХ СВЯЗЕЙ
Модифицирование поверхности за счет образования химических связей позволяет добиваться существенно большей устойчивости получаемых материалов к различным воздействиям внешней среды. При химическом закреплении можно снизить количество наносимого на поверхность активного компонента при сохранении, а иногда даже при улучшении характеристик продукта.
Рассматриваемые методы модифицирования делятся на две группы: «иммоблизация» и «сборка на поверхности». Метод иммобилизации заключается в получении целевого продукта путем ковалентного закрепления на поверхности носителя заранее синтезированного модификатора. Основным достоинством этого метода является простота и возможность создания на поверхности значительной концентрации нанесенного вещества. Главное ограничение состоит в том, что во многих случаях синтез требуемых модификаторов сложен, а иногда и вообще невозможен.
Метод сборки на поверхности заключается в том, что относительно простые химические соединения, привитые тем или иным способом к поверхности, подвергают дальнейшим последовательным превращениям.
Независимо от применяемого метода структура модифицированного носителя, например, оксида, может быть представлена в виде (рис. 7.1):
При выборе модификатора к нему предъявляется ряд следующих требований: 1) элемент, образующий якорную группировку, должен быть не менее, чем двухвалентен, т.к. в противном случае он не сможет реагировать с поверхностью; 2) селективность реакции (протекает быстро и однозначно); 3) стабильность получаемой структуры; 4) доступность модификатора; 5) отсутствие токсичности (например, запрет на применение алкильных соединений олова или ртути).
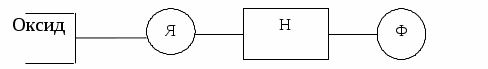
Рис. 7. 1. Структура неорганического оксида,
модифицированного органическими соединениями:
Я – якорная группа, ответственная за фиксацию на поверхности привитой молекулы; Н – «ножка», группировка, обеспечивающая связь между якорем
и функциональной группой Ф
Метод иммобилизации. Метод иммобилизации представляет собой одностадийный процесс модифицирования с использованием соединений, содержащих группировки, способные к образованию химических связей с поверхностью. Основными преимуществами данного метода являются простота, значительное количество привитого целевого вещества и однородность сорбционных центров.
Использование метода требует предварительной подготовки поверхности. Например, при модифицировании кремнезема его поверхность должна быть полностью гидроксилирована, что обычно достигается путем кипячения в воде в течение 24 часов. Затем физически адсорбированную воду удаляют с поверхности носителя путем обработки при давлении 1,3 Па и температуре 200 0С в течение 6–8 часов. Непосредственно модифицирование проводят с использованием растворов модификаторов в абсолютированных органических растворителях при слабом перемешивании и температуре не менее 95 0С, но на 5–10 0С ниже температуры кипения растворителя в течение не менее 8 часов. Полученный модифицированный кремнезем необходимо затем тщательно очистить экстракцией абсолютированными, неполярными, а затем и полярными растворителями, включая водно-органические смеси, и высушить.
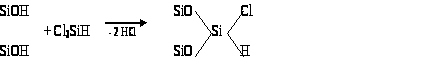
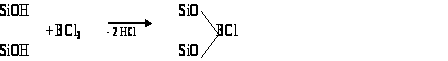
В качестве модификаторов при иммобизизации на поверхности используют кремнийорганические соединения, в основном хлор- или алкоксисиланы. Высокой реакционной способностью обладают также силозаны и силаны, содержащие у атома Si группу RCOO- или MeCOCH=C(Me)O–. Повышения светочувствительности оксидных материалов можно добиться путем иммобилизации на поверхности дипиридильных комплексов рутения Ru(dp)3. Широко применяется модифирование полимерами, для которого используют три типа полимеров: проводящие, ионообменные или редокс-полимеры. К проводящим полимерам относятся полиацетилен, полипиррол, полианилин, политиофен,паолипарафенилен и др.; ионообменными полимерами являются, например, перфторсульфонаты, полисилоксан, полииндол и др. . В качестве редокс-полимеров для модифицирования поверхности могут использоваться комплексы с переносом заряда, например полистирол-бипиридил-рутениевый комплекс, полимерные виологены, полимеры аминокислот, несущих редокс-группы.9
Модифицирование неорганическими веществами широко используется не только для получения новых материалов-адсорбентов и катализаторов, но и с целью изучения расположения и активности различных групп на поверхности носителей, в частности, оксидов. Значительный вклад в исследование процессов химического модифицирования минеральных веществ, в том числе кремнеземов, неорганическими соединениями внесла школа проф. В. Б.
Алесковского. Он с сотрудниками подробно изучил реакции модифицирования кремнезема галогенидами переходных металлов. Так, было установлено, что при модифицировании кремнезема при 180 0С TiCl4 соотношение Ti:Cl равно примерно 1,0, что указывает на образование поверхностных SiO3–TiCl – групп.
| |