|
|
Общие сведения о высокомолекулярных соединениях
Кинетика радикальной полимеризации в общем случае весьма сложна; дело в том, что она неоднородна; кинетические характеристики системы весьма значительно меняются с ростом глубины процесса. Причина, прежде всего, в том, что с увеличением степени конверсии мономера обычно значительно возрастает вязкость системы и уменьшается скорость диффузии крупных молекул (гель-эффект, см. ниже). Кроме того, по мере накопления полимера возрастает вероятность передачи цепи на полимер, осложняющая картину.
Однако при малых степенях конверсии мономера (не выше 10%) кинетика процесса достаточно простая; на ее основе можно сделать вполне определенные выводы. Далее будет рассмотрена именно этот вариант – кинетика при малых глубинах процесса (ее можно назвать элементарной кинетикой радикальной полимеризации).
Вначале рассмотрим наиболее простой случай, когда можно пренебречь реакциями передачи цепи; такой случай реален, если в реакционной смеси отсутствуют примеси, на которые может идти передача и если мономер не аллильный (тогда реакциями передачи цепи на мономер можно пренебречь). В этом случае можно считать, что протекают только реакции инициирования, роста и обрыва цепей.
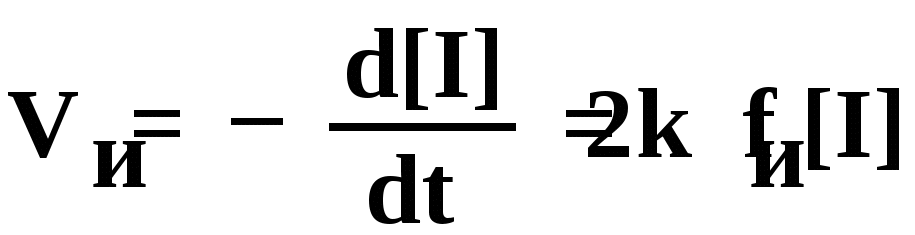
Скорость инициирования определяется скоростью распада инициатора (эта стадия лимитирующая, см. стр. 13); тогда:
г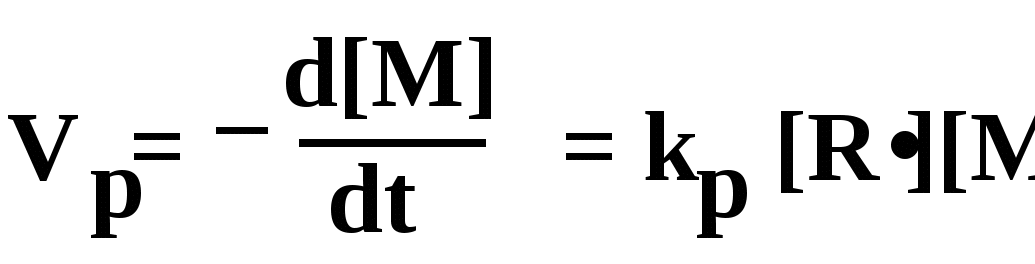
де vи – скорость инициирования, [I] – концентрация инициатора, kи – константа скорости иницирования, f – эффективность инициатора (стр. 15); множитель 2 отражает образование двух радикалов из молекулы инициатора (наиболее частый вариант)
Скорость роста цепи можно выразить уравнением:
где vр - скорость роста цепи, kр – константа скорости роста цепи, [M] –концентрация мономера, [R•] – концентрация радикалов (“живых» цепей).
Это уравнение отражает то, что любая реакция роста цепи – взаимодействие радикала с мономером (стр. 15). Оно справедливо при допущении, что константа роста kр не зависит от величины радикала R• (это допущение корректно).
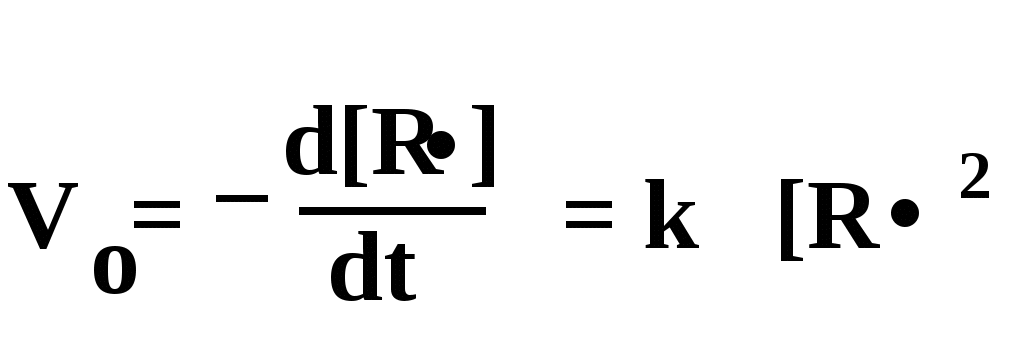
Скорость обрыва цепи выражается уравнением:
где vо - скорость обрыва цепи, kо – константа скорости обрыва цепи
Это уравнение отражает то, что обрыв происходит при взаимодействии двух радикалов («живых» цепей) (стр. 16).
Общая скорость полимеризации – это скорость расхода мономера (– d[M]/dt) и, следовательно, она равна скорости роста цепи
vпол = vp
Уравнение скорости роста цепи включает концентрацию радикалов [R•], которую трудно измерить. Однако концентрацию радикалов можно исключить из уравнения скорости роста, если допустить, что в ходе процесса концентрация радикалов постоянна. Это допущение называется условием квазистационарности; на начальных стадиях процесса (при небольших глубинах) оно выполняется хорошо. При таком допущении скорость образования радикалов равна скорости их исчезновения. Поскольку радикалы образуются на стадии иницирования, а исчезают на стадии обрыва, скорости этих реакций равны, т.е. vи = vо, т.е.:
Т
аким образом, скорость полимеризации пропорциональна концентрации мономера и корню квадратному из концентрации инициатора.
Средняя степень полимеризации (определяющая молекулярную массу полимера) в первом приближении равна длине кинетической цепи (стр. 17), т.е. соотношению скоростей реакций роста и обрыва цепи:
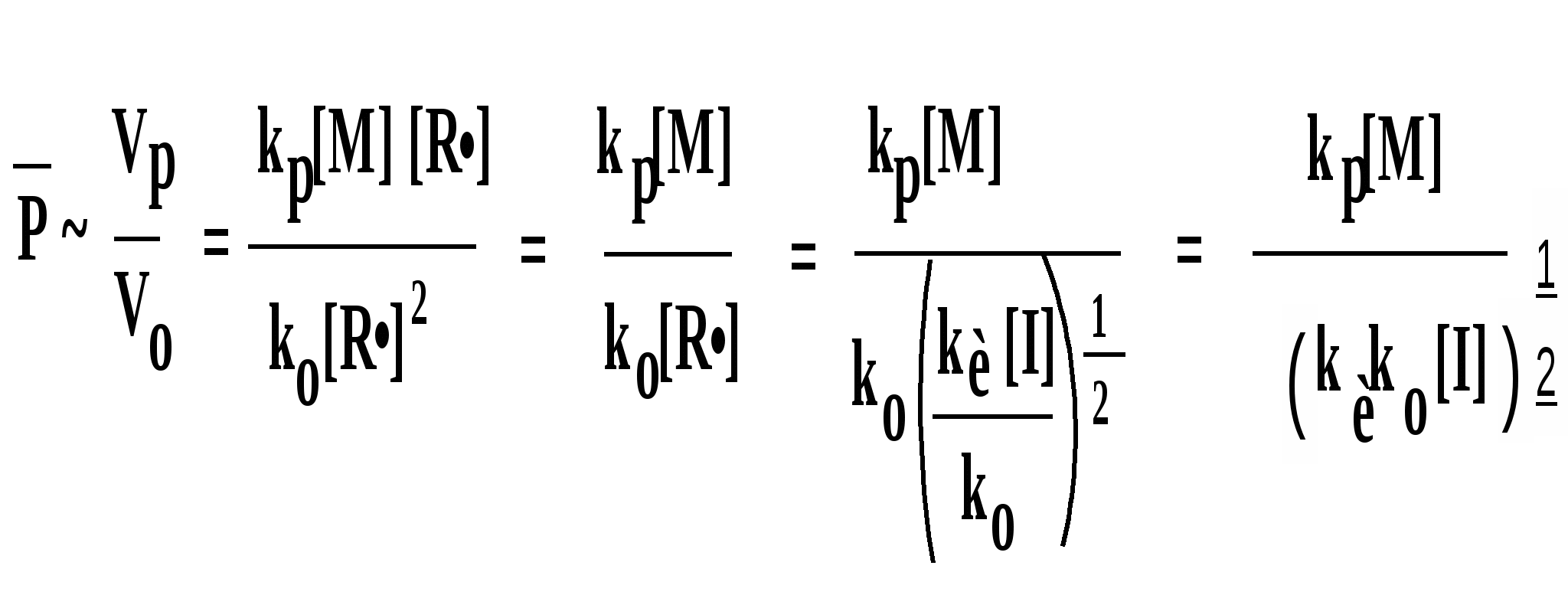
Таким образом, молекулярная масса полимера пропорциональна концентрации мономера и обратно пропорциональна корню квадратному из концентрации инициатора.
Итак, увеличение концентрации мономера ведет к увеличению как скорости полимеризации, так и молекулярной массы полимера, в то время как увеличение концентрации инициатора, увеличивая скорость процесса, снижает молекулярную массу. Последнее нетрудно понять и чисто качественно, т.к. при увеличении концентрации инициатора возрастает и концентрация растущих цепей, что увеличивает вероятность их встречи и обрыва цепей.
Теперь несколько усложним систему и учтем реакции передачи цепи (кроме передачи на «мертвый» полимер, так что рассматриваем по-прежнему кинетику при малых глубинах полимеризации). Обычно наибольшее значение имеют реакции передачи цепи на посторонние молекулы, прежде всего на регуляторы; ограничимся этим типом передачи.
Как уже указывалось, передача цепи на регулятор не влияет на скорость процесса. Средняя степень полимеризации (Pr) в этом случае равна (в первом приближении) отношению скорости роста цепи к сумме скоростей обрыва и передачи цепи (т.к. при передаче обрываются молекулярные цепи):

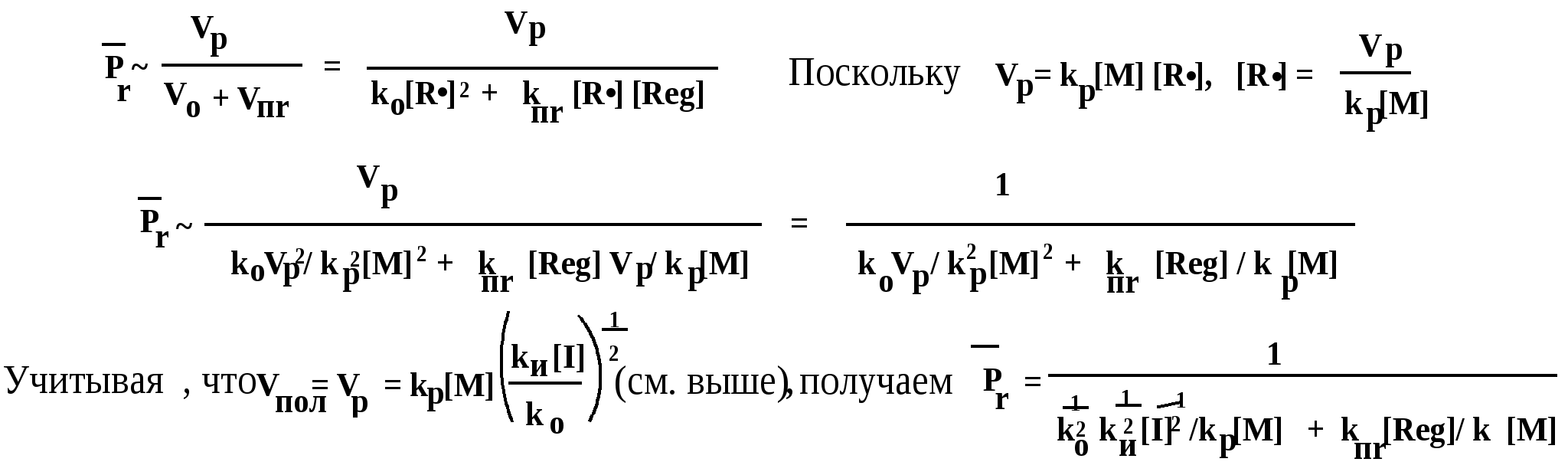
Приведенный выше анализ элементарной кинетики позволил определить зависимость скорости полимеризации и молекулярной массы полимера от концентрации мономера и инициатора, а для молекулярной массы – также от концентрации регулятора (если он присутствует). Кроме этого, на ход и результаты полимеризации влияет ряд других факторов, которые рассмотрены ниже.
Влияние температуры. А.В наиболее распространенном варианте полимеризации с участием инициаторов увеличение температуры приводит к увеличению скорости полимеризациии уменьшению молекулярной массы полимера. Увеличение скорости в комментариях не нуждается; уменьшение молекулярной массы связано с тем, что при повышении температуры скорость инициирования растет в большей степени, чем скорость роста цепи (т.к. инициирование имеет бòльшую энергию активации). Следовательно, по условию квазистационарности, и скорость обрыва цепи растет быстрее скорости роста, т.е уменьшается отношение vp/vo , а, следовательно, и молекулярная масса.
Б. При фотохимическом инициировании с ростом температуры увеличиваются и скорость процесса и молекулярная масса полимера. Это связано с тем, что с ростом температуры скорость фотохимического инициирования практически не меняется, а скорость роста цепи растет.
Другие следствия повышения температуры (для всех вариантов полимеризации): 1) повышение температуры уменьшает регулярность строения макромолекул полимеров, т.к. при этом возрастает вероятность сочленения элементарных звеньев по схемам «хвост к хвосту» и «голова к голове» (стр. 16); 2) Полимеризация винильных мономеров ( и диенов) – реакция экзотермическая (см. ниже); следовательно, при повышении температуры равновесие мономер полимер сдвигается влево; иными словами растет роль реакций деполимеризации. Все это не позволяет сколько-нибудь эффективно проводить радикальную полимеризацию при температурах выше 120 оС.
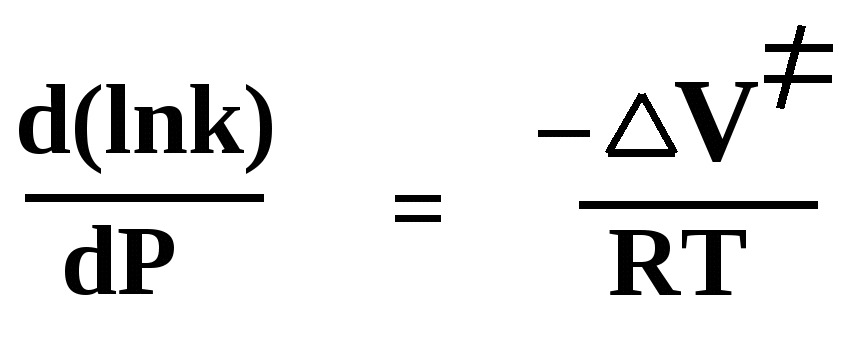
Влияние давления. Влияние давления (Р) на скорость любой химической реакции выражается уравнением Эванса – Поляни:
где k – константа скорости реакции, ΔV≠ - изменение объёма при образовании активированного комплекса (переходного состояния) из реагирующих частиц.
При радикальной полимеризации на стадии роста цепи ΔV≠ < 0, т.к. реакции роста цепи – бимолекулярные, а в таких реакциях объём при образовании переходного состояния уменьшается; следовательно, с увеличением давления скорость роста цепи (а, следовательно, и полимеризации в целом) увеличивается. Напротив, для реакции инициирования ΔV≠ > 0, т.к. здесь лимитирующая стадия – распад инициатора – мономолекулярная реакция, а в таких реакциях при образовании переходного состояния объём увеличивается. Следовательно, с ростом давления скорость инициирования, а значит и скорость обрыва цепи (по условию квазистационарности) уменьшается. Таким образом, растет соотношение vp/vo,, т.е. молекулярная масса полимера.
Полимеризация при высоких давлениях (порядка 1000 атм.) используется для этилена (образуется полиэтилен высокого давления).
Влияние глубины протекания процесса (степени конверсии мономера).
Влияние этого фактора наиболее сложно и сильно зависит от других условий проведения процесса.
А. В большинстве случаев при малых глубинах протекания процесса (примерно до 10%) скорость процесса и молекулярная масса полимера практически не меняются. Однако при увеличении глубины процесса наблюдается увеличение как скорости процесса, так и молекулярной массы полимера. Это может на первый взгляд показаться неожиданным, т.к. с ростом степени конверсии мономера уменьшается его концентрация, что, согласно приведенным выше кинетическим уравнениям (стр. 24) должно вести к уменьшению и скорости и молекулярной массы. Однако здесь кинетика уже совсем иная, в частности, не действует условие квазистационарности. Дело в том, что по мере накопления макромолекул полимера быстро возрастает вязкость системы (растворы полимеров, как известно, обладают исключительно высокой вязкостью, причем тем большей, чем выше их концентрация и молекулярная масса полимера). Возрастание вязкости приводит к резкому уменьшению подвижности больших частиц, в частности, «живых цепей», а, значит, и вероятности их встречи, т.е. обрыва цепи (обрыв цепи становится диффузионно-контролируемым процессом). В то же время подвижность малых частиц (молекул мономера) в довольно большом диапазоне вязкости системы сохраняется, так что скорость роста цепи не меняется. Резкое увеличение соотношения vp/vo приводит к значительному росту молекулярной массы полимера. Скорость распада инициатора, как мономолекулярной реакции, от вязкости не зависит, т.е. скорость образования радикалов выше скорости их исчезновения, концентрация радикалов растет, условиеквазистационарности не соблюдается.
Рассмотренные выше изменения, связанные с ростом вязкости, носят название гель-эффекта (иногда его называют также эффектом Тромсдорфа). При дальнейшем увеличении глубины процесса вязкость может возрасти настолько, что теряют подвижность и малые частицы; это приводит к замедлению реакции роста цепи, а затем и к ее полной остановке, т.е. к прекращению полимеризации. Гель-эффект особенно сильно проявляется при полимеризации в блоке (полимеризации чистого мономера); в достаточной степени проявляется он и при полимеризации в достаточно концентрированных растворах.
Б. Если проводить полимеризацию в сильно разбавленных растворах и при этом образуются полимеры с относительно невысокой молекулярной массой или если образовавшийся полимер выпадает из раствора, то вязкость в ходе процесса меняется мало; в этом случае гель-эффект не наблюдается, скорость процесса и молекулярная масса полимера меняются мало.
В относительно недавнее время изучены процессы полимеризации в присутствии специфических инициаторов; при этом молекулярная масса полимера относительно равномерно растет с увеличением глубины процесса.
Эти специфические инициаторы – ди- или полипероксиды и инифертеры.
Первые из них содержат две или более пероксидных группы в молекуле. При использовании этих инициаторов процесс протекает следующим образом (на примере инициатора с двумя пероксидными группами):
П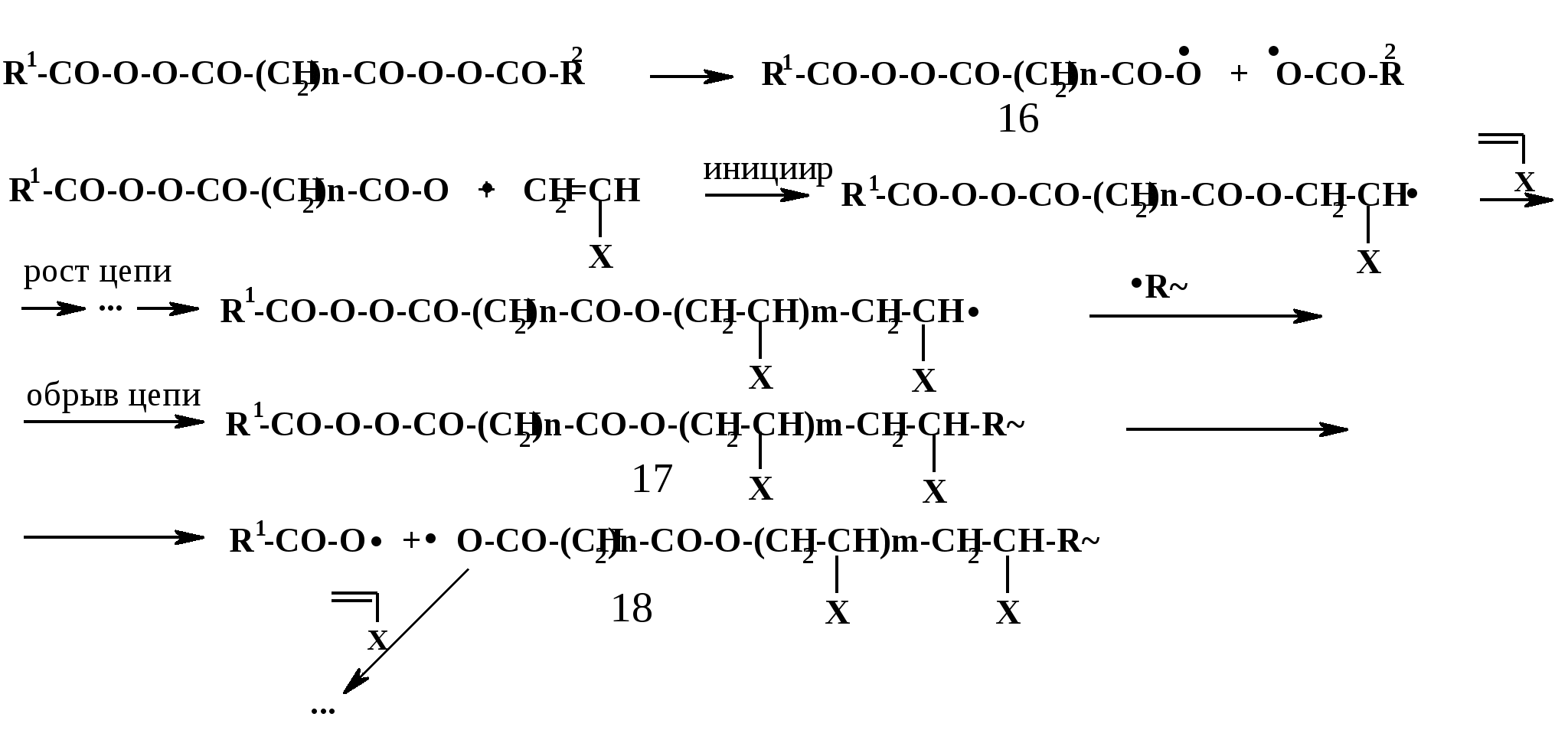
осле распада такого бис-пероксида образуются радикалы, один из которых (16) содержит пероксидную группировку. Радикал (16) инициирует рост полимерной цепи; затем происходит обрыв цепи при взаимодействии с другой «живой» цепью (обозначенной на схеме как •R) и образуется «мертвый» полимер (17). Этот полимер содержит лабильную пероксидную группу; в условиях проведения процесса эта группа распадается, образуя полимерный радикал (18), который начинает «достраиваться», реагируя с молекулами мономера; далее ситуация может повториться. Таким образом, по мере протекания процесса величина макромолекул постоянно растет.
Инифертеры – своеобразные соединения, которые являются не только инициаторами, но также активно участвуют в процессах передачи цепи и обрыва цепи; отсюда и их название, скомбинированное из некоторых букв английских названий этих реакций (Initiation – инициирование, Transfer – передача, Termination – обрыв цепи). Главная особенность этих инициаторов: при распаде они образуют два радикала, из которых только один активный, а второй – малоактивный – он не может инициировать рост полимерной цепи.
Одним из таких инифертеров является S-бензил-N,N-диэтилдитиокарбамид (19). В его присутствии происходят следующие реакции:
И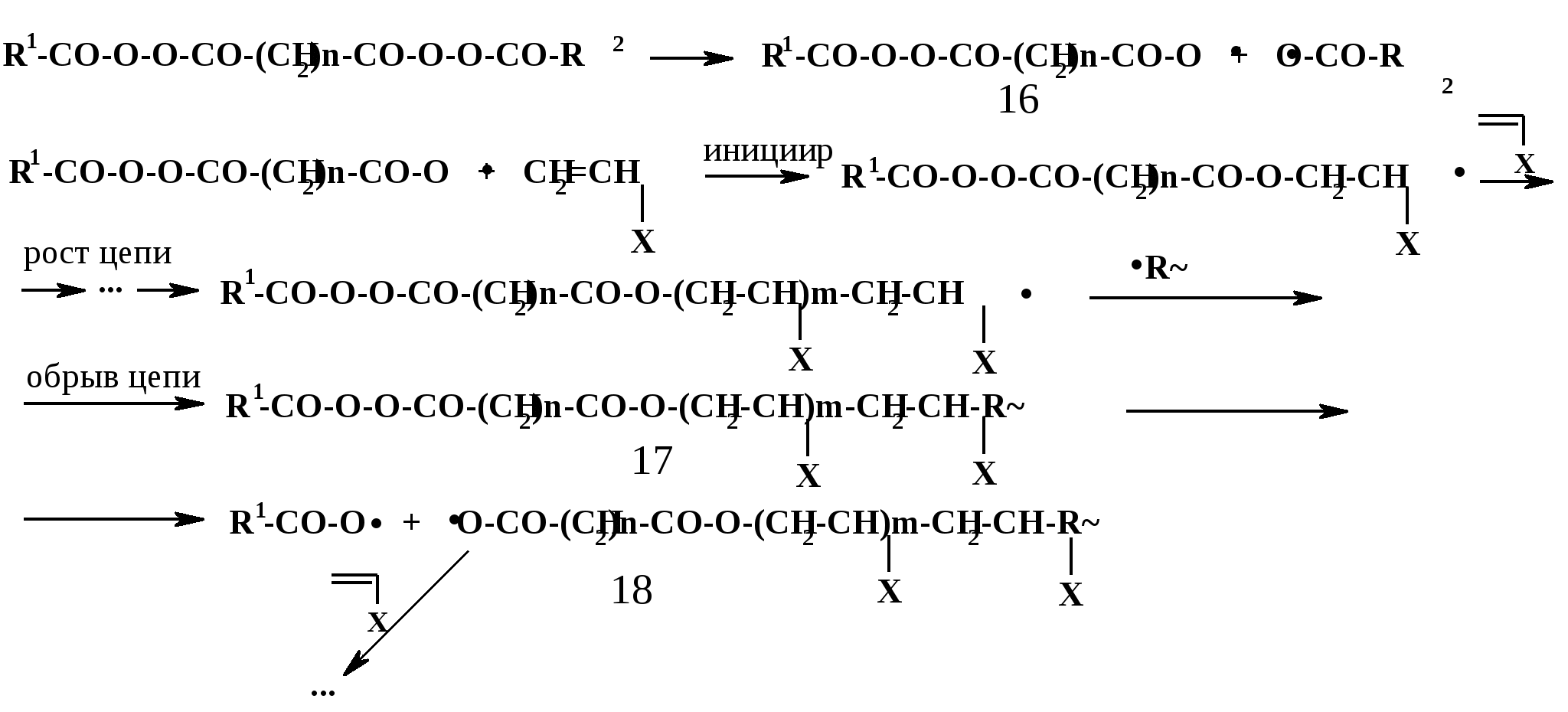
нифертер (19) распадается с образованием активного радикала (20) и неактивного радикала (21). Радикал (20) инициирует рост полимерной цепи. Растущая «живая» цепь может: А) передавать цепь на инициатор; Б) обрываться путем рекомбинации с неактивным радикалом (21); такая рекомбинация достаточно вероятна, потому что неактивные радикалы могут накапливаться в довольно значительной концентрации. И при передаче и при обрыве «живая» цепь превращается в один и тот же «мертвый» полимер (22), который содержит лабильные концевые звенья
CH2-CH(X)-S(C=S)-NEt2; эти звенья легко диссоциируют на радикалы по реакции, обратной рекомбинации, и «мертвый» полимер снова «оживает» и способен к дальнейшему росту. Поэтому и здесь молекулярная масса растет с увеличением глубины конверсии.
Процессы полимеризации в присутствии полипероксидов и инифертеров позволяют получать полимеры с меньшей степенью полидисперсности, чем процессы в присутствии обычных инициаторов; это положительно сказывается на их технических свойствах.
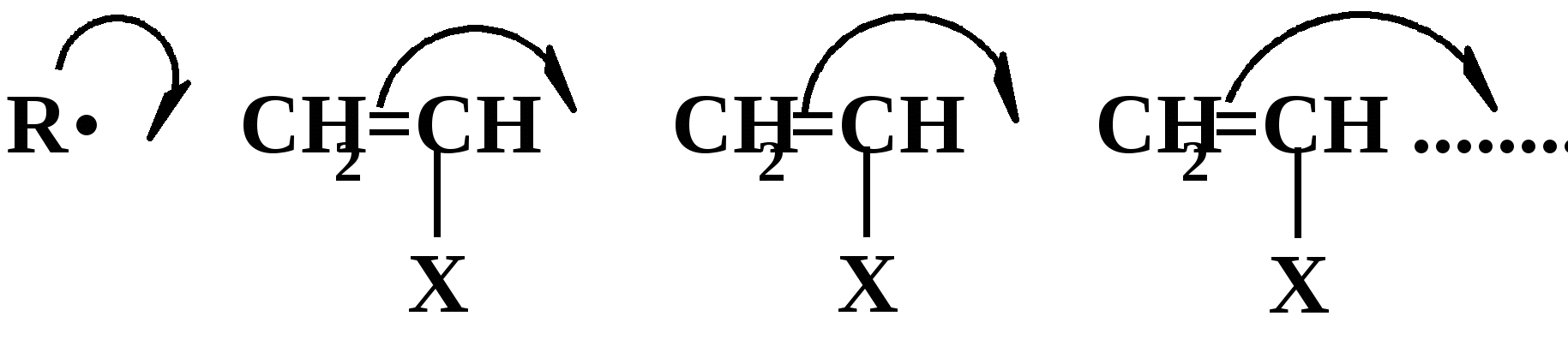
Влияние предварительной ориентации молекул мономера.Известно, что столкновение реагирующих частиц будет эффективным, если они ориентированы определенным образом. Если молекулы мономера перед началом полимеризации линейно ориентированы относительно друг друга:
то скорость роста цепи должна значительно возрасти, т.к. при каждой реакции роста радикал ориентирован точно на «голову» мономера, и практически каждое столкновение радикал – мономер будет эффективным (увеличивается значение фактора А в уравнении Аррениуса). Скорость обрыва цепей при этом не увеличивается, так что растет не только скорость полимеризации, но и молекулярная масса полимера.
Предварительная ориентация молекул мономера может быть достигнута, например, при полимеризации в соединениях включения (клатратах), когда молекулы мономера линейно ориентированы в каналах кристаллов соединения – «хозяина». Другие варианты – твердофазная полимеризация монокристаллов некоторых мономеров или полимеризация в мономолекулярных слоях на границе раздела фаз; эти варианты будут рассмотрены позднее, в разделе «Практические способы проведения полимеризации»
Радикальная сополимеризация
Все описанные выше закономерности были рассмотрены на примерах полимеризации одного мономера (гомополимеризации). Но, как известно, широко используется и сополимеризация – совместная полимеризациядвух или трех мономеров. Она проводится для получения полимеров с более широким спектром свойств, для получения материалов с заранее заданными свойствами, а также в фундаментальных исследованиях для выяснения реакционной способности мономеров. Продуктами сополимеризации являются сополимеры.
В принципе механизм радикальной сополимеризации вполне аналогичен механизму радикальной гомополимеризации. Однако здесь возникает несколько проблем.
1) Возможность сополимеризации – будут ли включаться в полимерную цепь звенья обоих (или трех) полимеров, или каждый мономер будет полимеризоваться отдельно и образуется смесь гомополимеров.
2) Соотношение между составом сополимера и составом взятой для процесса смеси мономеров. Здесь имеется в виду дифференциальный состав сополимера, т.е. его состав в данный момент (если брать интегральный состав, т.е. состав всей массы сополимера, то ясно, что при большой глубине процесса он примерно совпадет с составом смеси мономеров, однако при разных глубинах процесса могут образовываться макромолекулы с разным соотношением мономерных звеньев).
Если дифференциальный состав сополимера совпадает с составом взятой для полимеризации мономерной смеси, то сополимеризацию называют азеотропной. К сожалению, случаи азеотропной сополимеризации достаточно редки; в большинстве случаев дифференциальный состав сополимера отличается от состава смеси мономеров. Это означают, что в процессе полимеризации мономеры расходуются не в той пропорции, в которой они взяты; один из них расходуется быстрее другого, и по ходу реакции его необходимо добавлять для поддержания постоянного состава смеси мономеров. Отсюда ясно, сколь важно не только качественное, но и количественное решение этой проблемы.
3) Характер структуры получаемого сополимера, т.е. образуется ли статистический, чередующийся или блок-сополимер (см. стр. 7-8).
Решение всех этих проблем вытекает из анализа кинетики формирования макромолекулы сополимера, т.е. стадии роста цепи при сополимеризации (т.к. макромолекула сополимера образуется именно на этой стадии).
Рассмотрим наиболее простой случай сополимеризации двух мономеров, условно обозначив их символами А и В. Стадия роста цепи в этом случае, в отличие от гомополимеризации, включает элементарные реакции не одного, а четырех типов: действительно, в ходе роста образуются «живые» цепи двух типов – с концевым радикальным звеном мономера А [
A•, допустим,
CH2–CH(X)•] и с концевым радикальным звеном мономера В [
B•, допустим
CH2–CH(Y)•] и каждый из них может присоединяться к «своему» и «чужому» мономеру:
Дифференциальный состав сополимера зависит от соотношения скоростей этих четырех реакций, константы скоростей которых обозначены как k11…k21.
Мономер А входит в состав сополимера по реакциям 1) и 4); поэтому скорость расходования этого мономера равна сумме скоростей этих реакций:
М
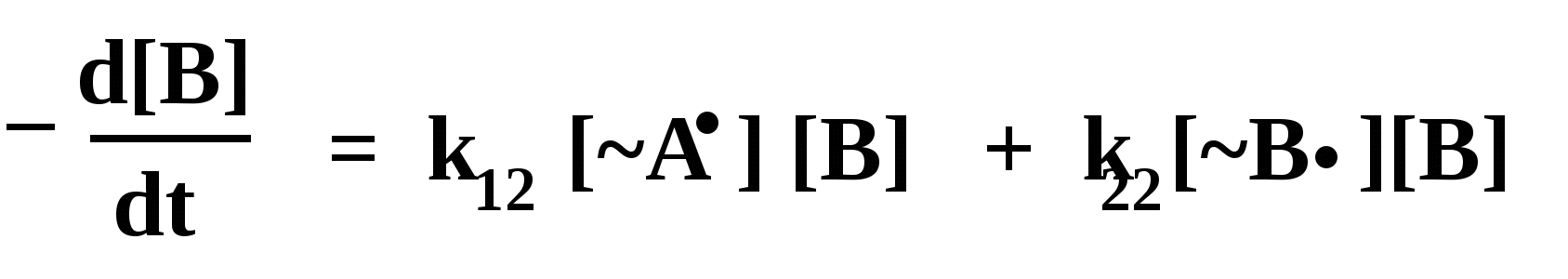
ономер В входит сополимер по реакциям 2) и 3), и для него:
Дифференциальный состав сополимера равен отношению скоростей вхождения в сополимер обоих мономеров:
В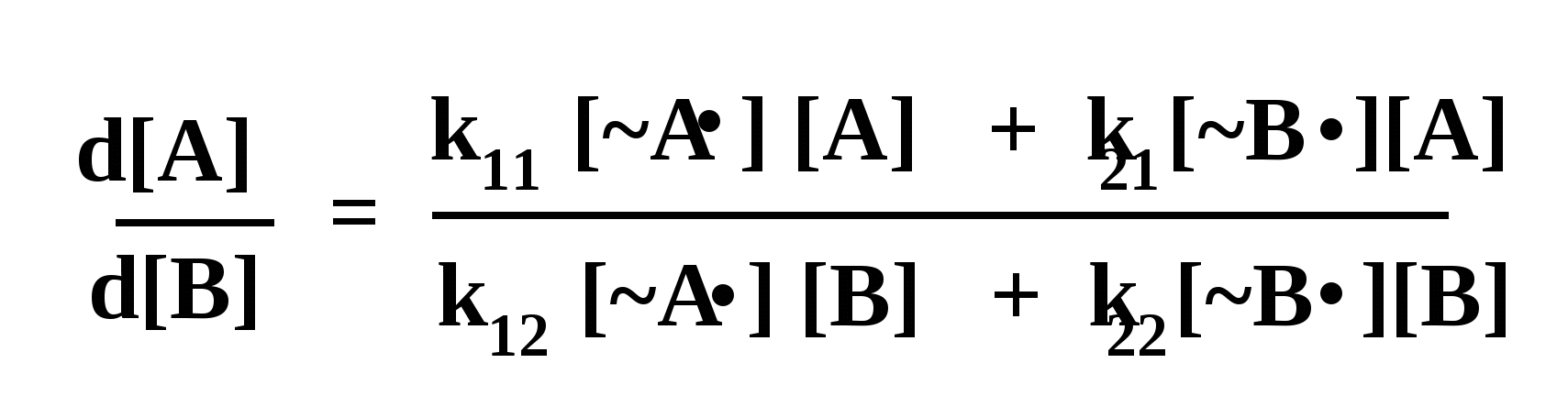
это уравнение входят трудно определяемые концентрации радикалов. Их можно исключить из уравнения, если ввести условие квазистационарности: концентрации обоих типов радикалов (
A• и
B•) постоянны; как при гомополимеризации, условие квазистационарности выполняется только при малых глубинах процесса. Из этого условия следует, что скорости взаимного превращения обоих типов радикалов одинаковы. Поскольку такие превращения происходят по реакциям 2 и 4, то:
П
одставляя полученное выражение для [A•] в уравнение дифференциального состава полимера, мы сокращаем в нем [B•] и, после ряда преобразований получаем:
Э
то уравнение носит название уравнения Мейо-Льюиса (иногда его называют уравнением Мейо). Это уравнение отражает зависимость дифференциального состава сополимера от состава мономерной смеси и от величин r1 и r2. Параметры r1 и r2 называются константами сополимеризации. Физический смысл этих констант вытекает из их определения: каждая из них выражает сравнительную активность каждого из радикалов по отношению к «своему» и «чужому» мономеру (константа r1 – для радикала
A•, константа r2 – для радикала
B•). Если радикал легче присоединяется к «своему» мономеру, чем к «чужому», ri > 1, если легче к «чужому», ri < 1. Иначе говоря, константы сополимеризации характеризуют сравнительную реакционнную способность мономеров.
Левая часть уравнения Мейо-Льюиса – дифференциальный состав сополимера. В правой части можно выделить два сомножителя: 1) состав мономерной смеси [A]/[B]; 2) сомножитель, включающий константы сополимеризации r1[A] + [B]/r2[B] + [A] = D (обозначим его символом D). Легко заметить, что при D=1 d[A]/d[B] = [A]/[B], т.е. сополимеризация азеотропна. Как уже упоминалось выше, случаи азеотропной сополимеризации достаточно редки, т.е. в большинстве случaев D ≠ 1. Таким образом, сомножитель D и есть тот фактор, который определяет отличие дифференциального состава сополимера от состава смеси мономеров. Если D > 1, то сополимер обогащен мономером А по сравнению с исходной смесью (т.е. мономер А расходуется в большей пропорции, чем мономер В). При D < 1, напротив, быстрее расходуется мономер В.
Величина сомножителя D полностью определяется величинами констант сополимеризации; следовательно именно константы сополимеризации определяют соотношение дифференциального состава сополимера и состава смеси мономеров, взятой для реакции.
Знание величин констант сополимеризации позволяет также судить о структуре полученного сополимера, а также о возможности или невозможности самой сополимеризации.
Рассмотрим основные варианты сополимеризации, определяемые величинами констант сополимеризации. Их удобно представить графически в виде кривых зависимости дифференциального состава сополимера от состава взятой для реакции смеси мономеров (рис. 3).
Р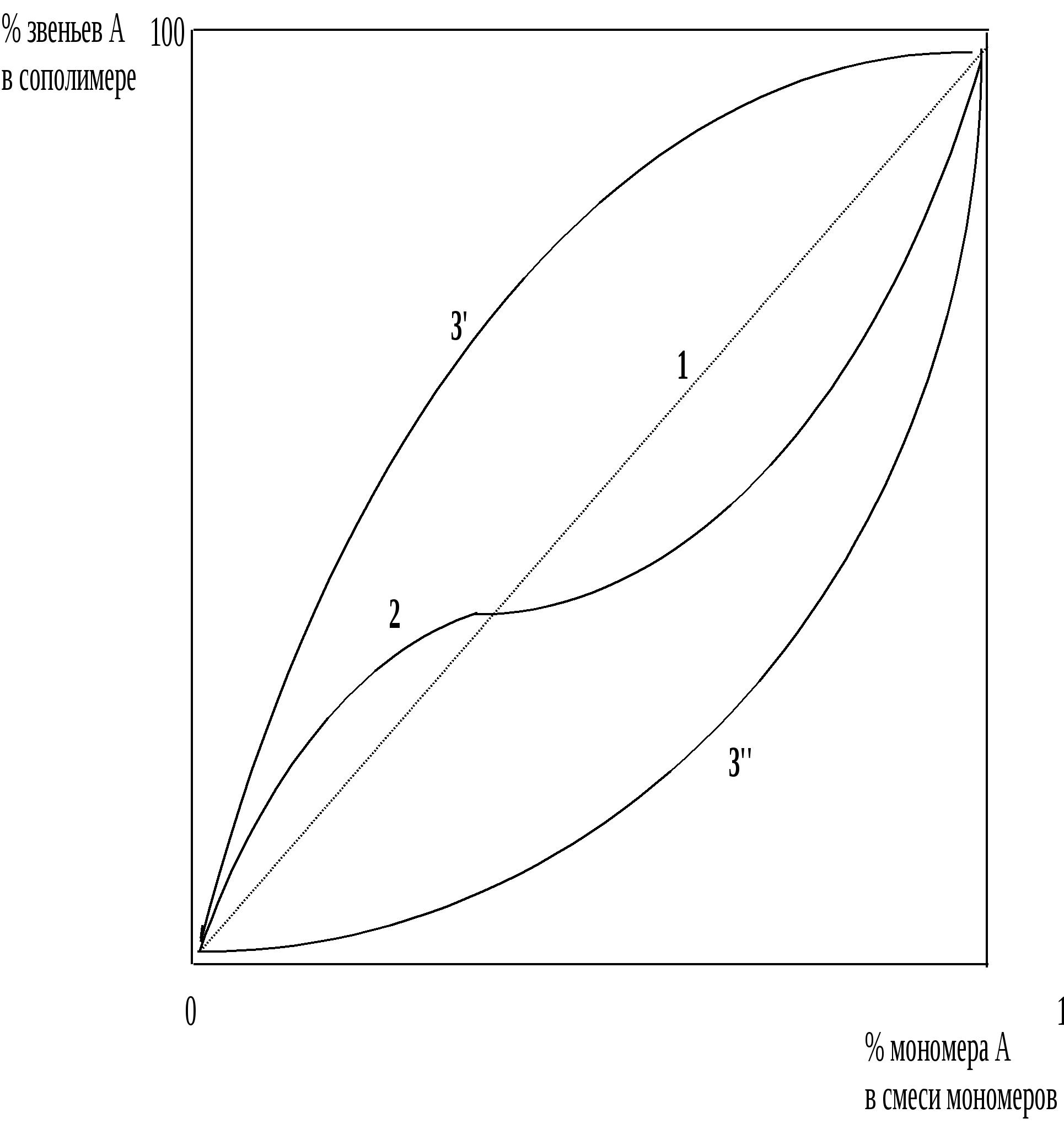
ис. 3. Зависимость дифференциального состава сополимера от состава смеси мономеров.
1. r1 = r2 = 1. В этом случае d[A]/d[B] = [A]/[B], т.е. при любом составе смеси мономеров происходит азеотропная сополимеризация. Это – редкий вариант. Графически он выражен пунктирной прямой 1 – линией азеотропа. Пример такой системы –сополимеризация тетрафторэтилена с хлортрифторэтиленом при 60 0С.
2. r1 < 1, r2 < 1 . Обе константы меньше единицы. Это означает, что каждый радикал предпочтительно реагирует с чужим мономером, т.е. можно говорить о повышенной склонности мономеров к сополимеризации.
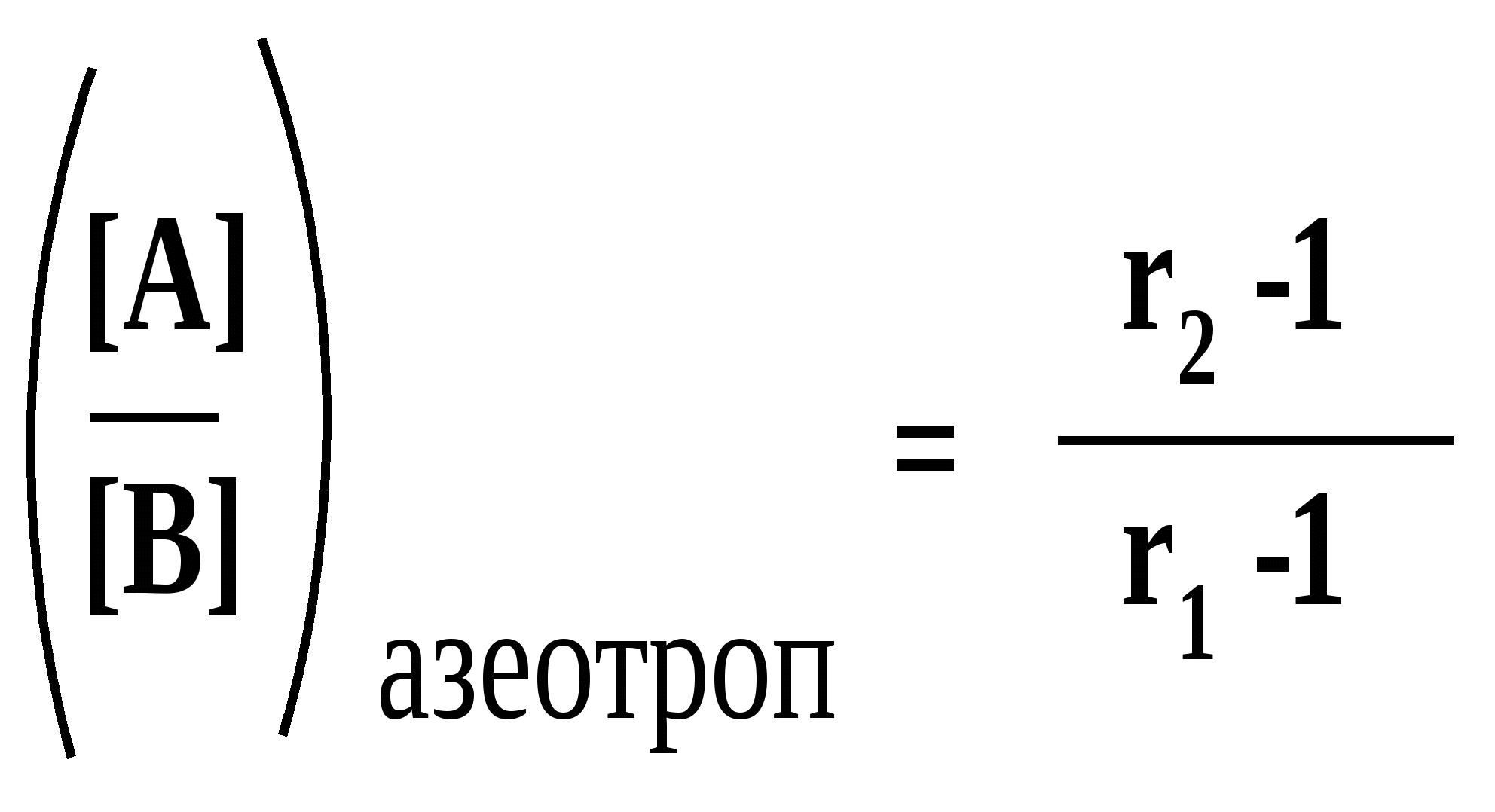
А) Состав сополимера. Дифференциальный состав сополимера обогащен тем мономером, которого мало в смеси мономеров (кривая 2 на рис. 3). Это легко вывести из анализа сомножителя D в уравнении Мейо-Льюиса: при [A] << [B] D < 1, следовательно, d[A]/d[B] < [A[/[B], а при [B] << [A] D >1 и d[A]/d[B] > [A[/[B]. Кривая 2 пересекает линию азеотропа, т.е. при каком-то одном соотношении мономеров полимеризация азеотропна. Это соотношение легко вычислить, т.к. в этом случае D = 1; отсюда:
Б) Структура сополимера. Поскольку каждый радикал предпочтительно присоединяется к чужому мономеру, в сополимере наблюдается тенденция к чередованию. Если константы сополимеризации не намного меньше единицы, эта тенденция выражена не очень значительно, и сополимер ближе к статистическому, чем к чередующемуся [коэффициент микрогетерогенности КМ (стр. 7) ближе к 1, чем к 2]. Но чем меньше величина констант, тем в большей степени структура полимера приближается к чередующейся. Предельный случай – бесконечно малая величина обеих констант (r 1 → 0, r 2 → 0); это означает, что каждый радикал реагирует только с «чужим» мономером, иначе говоря, каждый из мономеров в отдельности не полимеризуется, но вместе они образуют сополимер. Естественно, такой сополимер имеет строго чередующуюся структуру. Примером такой системы является пара: 1,2-дифенилэтилен – малеиновый ангидрид. Известны также случаи, когда одна из констант бесконечно мала, а другая имеет конечную величину; в таких случаях только один из мономеров сам не полимеризуется, но может образовывать сополимер со вторым партнером. Пример такой системы – стирол-малеиновый ангидрид.
3. r1 > 1, r2 < 1 или r1 < 1, r2 > 1 . Одна из констант больше единицы, другая – меньше единицы, т.е. один из мономеров легче реагирует со «своим» мономером, а второй – с «чужим». Это означает, что один из мономеров активнее другого в ходе сополимеризации, т.к. легче другого реагирует с обоими радикалами. Следовательно, при любом составе мономерной смеси дифференциальный состав сополимера обогащен звеньями более активного мономера (на рис. 3 – кривые 3’ для r1 > 1, r2 < 1 и 3’’ для r1 < 1, r2 > 1). Азеотропная полимеризация здесь невозможна.
Структура макромолекул сополимера в этом варианте наиболее близка к статистической. Частный (и не столь редко встречающийся) случай: r1r2 = 1, т.е. r1 = 1/r2, при этом величины констант не намного больше или меньше единицы. Это означает, что сравнительная активность мономеров по отношению к обоим радикалам одинакова (например, при r1 = 2, r2 = 0,5 мономер А в 2 раза активнее мономера В в реакциях как с радикалом
A▪, так и с радикалом
B▪). В этом случае способность каждого мономера к вхождению в полимерную цепь не зависит от природы радикала, с которым он сталкивается и определяется просто вероятностью столкновений с каждым из радикалов. Поэтому структура сополимера будет чисто статистической (КМ
1). Этот случай носит название идеальной сополимеризации – отнюдь не потому, что при этом образуется идеальный по свойствам сополимер (скорее наоборот), а по аналогии с понятием идеального газа, где, как известно, распределение частиц полностью статистическое. К наиболее известным примерам такой сополимеризации можно отнести сополимеризацию бутадиена со стиролом при 60 оС (r1 = 1,39, r2 = 0,78). В общем же случае вариант «одна константа больше единицы, другая меньше» – пожалуй, наиболее распространенный.
4. r1 > 1, r2 > 1. Обе константы больше единицы; каждый из радикалов предпочтительно реагирует со «своим» мономером; система обладает пониженной склонностью к сополимеризации. Что касается состава сополимера, то он должен быть обеднен тем мономером, которого мало в мономерной смеси. Эта картина прямо противоположна той, которая наблюдается для варианта r1 < 1, r2 < 1, а на рис. 3 была бы представлена кривой, зеркально подобной кривой 2. Но этот вариант сополимеризации встречается редко; можно разве что упомянуть сополимеризацию бутадиена с изопреном при 50 оС (r1 = 1,38, r2 =2,05), где константы лишь не намного больше единицы. Зато, к сожалению, встречаются случаи, когда обе константы бесконечно велики (r1→, r2); в этом случае сополимеризация просто не происходит, каждый из мономеров полимеризуется отдельно и образуется смесь двух гомополимеров (пример – пара: бутадиен – акриловая кислота). Весьма полезным был бы вариант, где константы имели бы большую, но конечную величину; в этом случае образовывались бы блок-сополимеры; к сожалению, таких случаев пока не найдено.
Термин «константы сополимеризации» нельзя воспринимать слишком буквально: их величины для данного мономера могут заметно меняться при изменении условий реакции, в частности, при изменении температуры. Например, при сополимеризации акрилонитрила с метилакрилатом при 50 оС r1 = 1,50, r2 = 0,84, а при 80 оС r1 = 0,50, r2 = 0,71. Поэтому, приводя значения констант, надо обязательно указывать условия.
|
|
|
 Скачать 0.64 Mb.
Скачать 0.64 Mb.