Учебник для медицинских и фармацевтических вузов Под ред. В. А. Черешнева и В. В. Давыдова. М. Издво, 2006. 1050 с
 Скачать 3.85 Mb. Скачать 3.85 Mb.
|

12.1. История изучения апоптозаТермин «апоптоз» (в переводе с греч. Означает опадание листьев с деревьев осенью) введен в научный обиход австралийским патологоанатомом Керром и соавт. в 1972 г. для обозначения формы гибели клеток, прототипом которой является гибель тимоцитов, возникающая под действием глюкокортикоидов. Хотя первое детальное описание смерти клеток как физиологического явления было дано в 1895 г. Флемингом, который впервые описал распад клеток овариального эпителия на частицы (впоследствии названные апоптозными или апоптотирующими тельцами), определив процесс быстрого исчезновения образовавшихся при распаде клеток фрагментов цитоплазмы и ядра как хроматолизис. Данная форма клеточной смерти была отождествлена с ранее описанной Куссманом (1950) программированной гибелью клеток эмбриона. В научной литературе чаще встречается термин апоптоз. ДеДювом (1953)была выдвинута лизосомальная концепция физиологической гибели клеток. Автор предложил, что при определенных условиях клетка может сама себя убить, используя для этой цели собственные лизосомы. Это явление получило название клеточного суицида. Однако вскоре была описана не связанная с лизосомами гибель клеток в результате накопления в ней свободных радикалов и свободного внутриклеточного кальция. Понятие же «апоптоз» было сформулировано Вилли только в 1980 г. Данное понятие было связанно с деградацией хроматина, катализируемой эндонуклеазами. Биохимически активный характер апоптоза подтвержден в лаборатории этого автора в 1984 г., где была показана значимость макромолекулярного синтеза для реализации процесса апоптоза. В 1987 г. автор сформулировал 4 основных элемента апоптоза: 1) уменьшение объема апоптотирующей клетки; 2) конденсация и фрагментация хроматина на ранних стадиях апоптоза с формированием так называемых апоптотических телец; 3) изменение мембраны апоптотирующей клетки, приводящее к распознаванию ее фагоцитами; 4) сопряженность апоптоза с активным белковым синтезом. Хотя еще в 1976 г. Скалка с соавт. и в 1979 г. К.П. Хансон, высказали мысль о том, что радиационная гибель лимфоцитов в интерфазе представляет собой проявление программированной их гибели, то есть апоптоза. Возникшее с конца 80-х годов большое внимание к апоптозу стало в последние годы XX века и первые годы XXI века всеобщим, особенно в связи с тем, что появились методические возможности регистрации различных проявлений апоптоза и анализа его молекулярных механизмов. Изучение апоптоза оказалось очень продуктивным для понимания ряда жизненно важных явлений и процессов, включая индивидуальное развитие организма, иммунный гомеостаз и онкогенез. В связи с развитием учения об апоптозе возникла необходимость пересмотра концептуальных основ не только физиологии, но и патологии. Вместо прежних представлений о гибели клеток многоклеточного организма как отрицательном биологическом явлении, идентифицируемом с некрозом, сформировался новый взгляд, согласно которому гибель части клеток в пределах организма является закономерным и необходимым гомеостатическим процессом, обусловленным апоптозом. Само существование многоклеточного организма подразумевает баланс жизни и смерти на уровне составляющих его клеточных популяций. В нормальных условиях в процессе жизни многоклеточного организма отмечаются два взаимоуравновешивающих процесса: пролиферация и гибель клеток, что обеспечивает сохранение постоянства клеточного состава данного организма. 12.2. Определение понятия «апоптоз» Апоптоз или запрограммированная (контролируемая) клеточная гибель представляет собой активную форму гибели клетки многоклеточного организма, являющуюся результатом реализации ее генетической программы в ответ на внешние или внутренние сигналы и требующую затрат энергии и синтеза макромолекул de novo. Морфологически апоптоз проявляется в уменьшении размера клетки, конденсации и фрагментации хроматина, уплотнении наружной и цитоплазматических мембран без выхода содержимого клетки в окружающую среду. 12.3. РОЛЬ АПОПТОЗА В ЖИЗНИ ЗДОРОВОГО ОРГАНИЗМА В многоклеточном организме апоптоз является повсеместно и постоянно происходящим процессом. Апоптоз играет ключевую роль в эмбриональном развитии, процессе метаморфоза, нормальном обновлении тканей. Благодаря апоптозу обеспечивается возрастная инволюция половых желез, тимуса, гепатоцитов, нейронов, костей, некоторые иммунные процессы и т.д. В частности, с помощью апоптоза из организма удаляются нежелательные или потенциально опасные клетки, например, аутореактивные Т-лимфоциты, инфицированные вирусами клетки, опухолевые клетки и т.д. Цитотоксические Т-лимфоциты и натуральные киллеры (НК) осуществляют элиминацию аллогенных и злокачественно трансформированных клеток путем индукции в них апоптоза. Апоптоз может нести ответственность и за ослабление или прекращение иммунных реакций. Доказана роль апоптоза в развитии старения. Нарушение процесса физиологической клеточной гибели может привести к развитию патологических состояний и заболеваний, проявлениями которых могут быть как дегенеративные, так и пролиферативные изменения. В частности, активация апоптоза является одним из патогенетических звеньев СПИДа, нейродегенеративных и миелодиспластических процессов, ишемических поражений тканей и органов. Ингибирование апоптоза лежит в основе развития опухолей, ряда аутоиммунных и вирусных заболеваний. В последние годы выявлена роль апоптоза как патогенетического фактора в развитии атеросклероза, инфаркта миокарда, травм, ишемии/реперфузии и др. Изучение роли апоптоза в развитии различных видов патологии может дать возможность его направленной коррекции и повысить тем самым эффективность лечения многих заболеваний. Тонкий, строго контролируемый баланс процессов апоптоза, пролиферации, дифференцировки и стабильного функционирования клеток (адгезии, передвижения и т.д.) рассматривается не только как ведущий механизм клеточного гомеостаза на уровне целостного многоклеточного организма, но и как главное условие его нормальной жизни. Показано, что роль апоптоза в популяциях неделящихся или слабо делящихся клеток минимальна или незначительна. В то же время в популяциях интенсивно делящихся клеток роль апоптоза огромна в силу того, что процесс апоптоза уравновешивает процессы пролиферации и дифференцировки клеток и поддерживает необходимую численность клеточной популяции. Принято считать, что интенсивность апоптоза наиболее высока в период эмбриогенеза. В последующие периоды онтогенеза она снижается и достигает минимума в старости, сохраняясь лишь в тканях с интенсивно делящимися клетками (гемопоэтическая ткань костного мозга, ворсинки слизистой тонкого кишечника и др.). Чем плотнее контакты между клетками и с межклеточным матриксом, тем слабее выражен их апоптоз, и наоборот. Установлено, что в обеспечении межклеточных контактов важная роль принадлежит мембранным ФАВ-интегринам. Среди ФАВ, защищающих клетки от апоптоза, следует назвать такие цитокины, как фактор роста фибробластов, коллаген II, фактор роста нервов, эритропоэтин, ИЛ-10, ИЛ-7 и многие другие, обеспечивающие жизненную активность соответственно фибробластов, хондроцитов, нейронов, эритроидных клеток, клеток миелоидного ряда, клеток лимфоидного ряда и мн. др. К цитокинам, способствующим развитию апоптоза, относятся фактор некроза опухолей -ФНО (TNF-), рост-трансформирующий фактор- (TGF), хемокин MIP-1, ИФН- и др. Роль апоптоза в жизни здорового организма заключается в:
12.3.1. Основные типы гибели клеток и их отличия Гибель клеток реализуется либо в виде некроза, либо апоптоза. Некроз клетки является результатом прямого цитотоксического воздействия на нее внешних повреждающих факторов и, как правило, сопровождается развитием воспаления. Некроз — это патологический процесс, возникающий в результате воздействия на организм патогенного агента. Апоптоз в нормальных условиях является физиологическим процессом, в основе которого лежит генетически запрограммированная гибель клетки. Апоптоз является реализацией закодированных в генотипе клеток суицидальных механизмов и не влечет за собой воспалительных реакций. То есть, некроз можно рассматривать как патологическую форму клеточной гибели, развивающуюся в результате острого клеточного повреждения, быстро приводящего к лизису клетки. Апоптоз — естественная, «физиологическая» форма гибели клетки, представляющая собой активный и строго контролируемый процесс самоуничтожения клетки. Между апоптозом и некрозом имеется ряд существенных различий. Апоптоз подвергается фармакологической коррекции, то есть его можно корригировать. При некрозе фармакологическая коррекция неэффективна. Последовательность морфологических изменений клетки при апоптозе и некрозе, согласно данным Хармон и соавт. (1990), М.А. Пальцева и соавт. (2003), представлена на рис. 12-1. При апоптозе наблюдаются следующая последовательность ультраструктурных изменений: Исходно интактная клетка (1). Уплотнение и сегрегация хроматина в ядре (2). Распад ядра на фрагменты и образование апоптозных телец (3). Фагоцитоз апоптозных телец соседней клеткой (4). При некрозе отмечается: Ранняя стадия, включающая конденсацию хроматина в нерезко очерченные массы и деградацию цитоплазматических структур (5). Разрушение мембран и дезинтеграция клетки (6). Апоптоз – довольно быстро протекающий процесс, занимающий обычно в культуре тканей несколько минут, а в организме – несколько часов. Самым ранним признаком апоптоза, выявляемым на электронно-микроскопическом уровне, являются резко очерченные уплотнения ядерного хроматина в виде гомогенной массы, а также некоторая конденсация (уплотнение) цитоплазмы. Затем ядро, молекулы ДНК и цитоплазма распадаются на фрагменты, причем цитоплазматические фрагменты разделяются цитоплазматической мембраной. Одним из главных отличительных признаков апоптоза является сохранность мембраны, а также фрагментов цитоплазмы, ядра и ДНК. 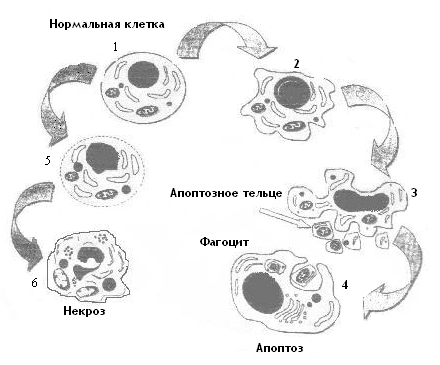 Рис. 12-1. Последовательность ультраструктурных изменений при некрозе (слева) и апоптозе (справа) Сохранение целостности цитоплазматической мембраны апоптотических телец позволяет, в отличие от некроза, избежать попадания содержимого клетки (лизосомальных гидролаз и других белков, цитокинов, калия, кальция) в межклеточное пространство и дает возможность макроорганизму сохранить толерантность к данному, постоянно идущему, процессу. Это обстоятельство особенно важно, если учесть, что в течение жизни в макроорганизме отмирает путем апоптоза астрономическое количество клеток (только масса кроветворных клеток составляет около 5 тонн). В результате апоптоза клетка превращается в совокупность окруженных цитоплазматической мембраной апоптотических телец, в которых плотно упакованные органеллы могут выглядеть интактными. В одних апоптотических тельцах ядерные компоненты отсутствуют, а в других — присутствуют (хроматин в них всегда очень плотный, резко очерчен и сконденсирован у ядерной мембраны). Апоптотические тельца быстро фагоцитируются окружающими соседними клетками (где они утилизируются с помощью лизосом). Эти клетки довольно быстро сближаются, при этом изменений цитоархитектоники тканей не происходит. Некоторые апоптотические тельца (например, в поверхностном эпителии) слущиваются. При апоптозе полностью отсутствуют признаки воспаления. Ультраструктурные проявления некроза значительно отличаются от характерной для апоптоза картины. Они сводятся, главным образом к сморщиванию органелл и дезинтеграции цитоплазмы. Хотя хроматин в некротизирующихся клетках так же, как и при апоптозе, конденсируется у ядерной мембраны, его компактные массы менее однородны и менее четко очерчены по краям ядра. При некрозе всегда, в отличие от апоптоза, происходит разрушение клеточных и внутриклеточных мембран. Повреждение мембран лизосом всегда приводит к высвобождению лизосомальных гидролаз и последующему распаду клетки. На более поздней стадии некроза хроматин из ядра исчезает в результате кариолизиса. Некроз обычно сопровождается развитием воспаления (чаще экссудативного). При некрозе, в отличие от апоптоза, восстановления цитоархитектоники ткани не происходит. Важно подчеркнуть, что на ранней стадии развития некроза возникающие изменения в виде нарушения ионно-обменной функции мембранных каналов и уменьшения содержания в клетке макроэргов потенциально обратимы. При нарастании этих изменений в клетке развиваются необратимые процессы. Основные отличия апоптоза и некроза представлены в таблице 12-1. Основные типы проявлений апоптоза Смерть клетки в процессе эмбриогенеза позвоночных. Несмотря на то, что апоптоз ответственен за реализацию многих морфогенетических процессов в процессе эмбриогенеза, гибель клетки в процессе раннего онтогенеза не является жестко детерминированной. Чтобы погибнуть, клетки должны заблаговременно получить сигнал к включению генетичесчкой программы апоптоза. Смерть клеток в интактных тканях взрослых особей. Апоптоз характерен как для медленно пролиферирующих клеточных популяций (гепатоцитов, клеток эпителия коры надпочечников), так и для быстро пролиферирующих (клеток кишечного эпителия, сперматогониев в период дифференцировки). В первом случае апоптоз уравновешивает процессы митоза, а во втором – большая часть митозов компенсируется не только апоптозом, но и в результате потери клеток за счет миграции. Гомеостатическая регуляция нормального количества и объема клеток осуществляется циклической продукцией факторов роста, стимулирующих митоз, и эндогенных «факторов смерти», индуцирующих апоптоз. Лимфоциты тимуса и других тканей, выполняющие определенные функции иммунной системы; мегакариоциты, распадающиеся на кровяные пластинки; стареющие гранулоциты, особенно нейтрофильные лейкоциты; Таблица 12-1 Основные морфологические и биохимические отличия апоптоза и некроза
желтое тело яичника, семенники и другие подвергающиеся инволютивным процессам структуры в процессе их старения также подвергаются разрушению путем апоптоза. Смерть клеток в процессе патологической атрофии и гиперплазии. Апоптоз лежит в основе развития патологической атрофии, особенно в эндокринно зависимых тканях. Например, благодаря апоптозу, после кастрации атрофируется предстательная железа. Кора надпочечников атрофируется после супрессии секреции АКТГ с помощью глюкокортикоидов. Патологическое разрастание тканей (за счет их гиперплазии) вызывается путем стимуляции митозов и торможения апоптоза. Возвращение же этих тканей к нормальному объему осуществляется путем стимуляции апоптоза. Альтруистический суицид клеток. Смерть клеток-мутантов и клеток, пораженных вирусом, может играть биологически полезную роль в их элиминации из организма. Выживание же этих клеток является вредным для организма в целом. Проникающая радиация вызывает апоптоз в популяции пролиферирующих клеток эпителия крипт кишечника, а не в пролиферирующих клетках лимфоидных органов, где лимфоциты, мутировавшие после облучения, могут стать причиной различных аутоиммунных заболеваний. Известно, что апоптоз стимулируется препаратами, применяемыми для химиотерапии опухолевых заболеваний. Уничтожение пораженных вирусами клеток путем апоптоза обеспечивает минимальное повреждение тканей, в то время как благодаря некрозу гибнет огромное число клеточно-тканевых структур. Клеточная смерть, вызванная минимальным повреждением. При слабом повреждающем воздействии на клетки активируется апоптоз, благодаря которому частично измененные клетки выбраковываются из популяции. При сильном повреждающем воздействии развивается некроз. Так, нагревание клеток в культуре тканей до температуры +43°-+44° С в течение тридцати минут вызывает апоптоз отдельных клеток, а гипертермия клеток, достигающая +46°-+47° С, приводит к массированному их некрозу. Апоптоз чаще всего идентифицируется морфологически (табл. 12-1). Критерии биохимической идентификации апоптоза хорошо изучены in vitro и плохо in vivo. Доказано, что в основе апоптоза лежит изменение ионного состава цитоплазмы клетки, приводящее к уменьшению содержания в ней внутриклеточного кальция, а также снижение содержания макроэргических фосфорных соединений. Генетический контроль физиологической клеточной гибели Роль генов в регуляции апоптоза, как и других физиологических процессов в клетке – пролиферации и дифференцировки, считается общепризнанной. В реализации (как запуске, так и модуляции) естественной клеточной гибели доказана роль генотипа, определяющего с участием различных генов последовательность биохимических и морфологических событий апоптоза, поглощения фагоцитами апоптотических телец, пикноз ядра, опзделение ядра на фрагменты, повреждение ДНК и др. Схематично роль генов в регуляции апоптоза представлена на рис. 12-2. Индукция и осуществление различных этапов апоптоза клеток различных тканей и органов контролируется набором генов, которые обуславливают выбор апоптотического пути, реализацию или ингибирование апоптоза, а также расщепление погибших клеток и их фагоцитоз. Доказано, что в модуляции апоптоза клеток принимают участие изменения активности следующих генов: p53, ras, Fas/APO-1, c-mys, c-jun, c-fos, nur77, bax, bcl-2, bcl-Х, be 12 и др. 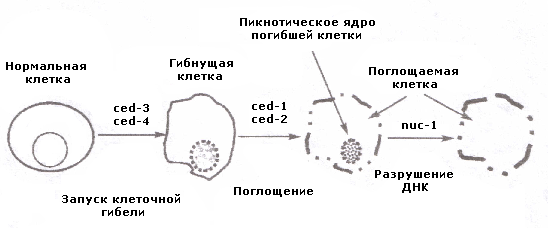 Рис. 12-2. Генетическая иерархия контроля клеточной гибели (Швартц, 1993) В развитии апоптоза, наряду с внутренней (генетической информацией), немаловажное значение придается влиянию на организм (его клеточные и субклеточные структуры) внешней информации в виде наличия или отсутствия, во-первых, растворимых сигнальных молекул, во-вторых, взаимодействия с другими клетками, в-третьих, взаимодействия с определенными субстратами. Пусковыеи внутриклеточныемеханизмы апоптоза В развитии апоптоза выделяют 3 стадии: индукторную, эффекторную и стадию деградации. Две последние стадии едины для всех разновидностей апоптоза. Первая стадия отличается большим разнообразием и существенно зависит от типа клеток и сигналов (индукторных факторов). Показано, что в одних случаях генетическая программа гибели клетки включается внешними факторами, а в других случаях эта программа реализуется при отсутствии соответствующих защитных факторов (табл. 12-2). Наиболее полно изучены условия экзогенной индукции (внешними воздействиями) апоптоза Т-лимфоцитов, особенно кортикальных тимоцитов. Классическими индукторами апоптоза тимоцитов служат глюкокортикоиды. Последние после проникновения в клетку проявляют свое действие через рецепторы, локализующиеся в ядре. Одни тимоциты гибнут под действием кор Таблица 12-2 Разновидности сигналов, приводящих к индукции апоптоза
тикостероидов, а другие – нет (являются гормонрезистентными). Сигнал, поступающий в Т-лимфоциты через рецептор для антигена, в норме может либо приводить к активизации пролиферации клеток, либо вызывать их апоптоз (обозначаемый как активационный). В качестве сигналов апоптоза могут служить: 1) отсутствие костимуляции через мембранные молекулы CD28, CD40 и др.; 2) отсутствие ростовых факторов, прежде всего ИЛ-2; 3) предварительное перекрестное сшивание молекул CD4; 4) мембранные молекулы лимфоцитов CD2; 5) молекулы главного комплекса гистосовместимости класса I; 6) 1- и 2-интегрины; 7) ФНО и т.д. Сигнал к развитию апоптоза подается, главным образом, через рецептор цитокина TNF-, который имеет цитоплазматический домен гибели, передающий летальный сигнал внутрь клетки. Некоторые цитокины, например ИЛ-2 и интерферон-, в зависимости от ситуации могут либо индуцировать, либо предотвращать развитие апоптоза. Такая амбивалентность эффектов свойственна и ряду других факторов: ингибиторам синтеза белка, активаторам и ингибиторам активности протеинкиназ и т.д. Существуют также рецепторы, для которых передача сигнала к развитию апоптоза является основной их функцией. Это Fas-рецептор (АРО-1, CD95) и белки группы DR (Death receptors — «рецепторы смерти»): DR3(АРО-3), DR4 (TRAIL-R1), DR5 (TRAIL-R2). В процессе передачи внутриклеточного сигнала, приводящего к развитию апоптоза, существуют две фазы. Первая, более ранняя фаза весьма вариабельна, ее характер зависит от вида пусковых механизмов (FasL → Fas, TNF → TNFR1 и т.д.). Следующая, вторая фаза универсальна для всех разновидностей апоптоза (рис. 12-3). Как уже указывалось выше, первая фаза передачи апоптогенного сигнала может быть различной в зависимости от вида сигнала, что иллюстрируют следующие примеры. Как видно на рис. 12-3, в случае Fas-зависимого апоптоза связывание Fas-лиганда (FasL) с тримерным Fas-рецептором приводит к конформационным изменениям в цитоплазматическом домене смерти рецептора Fas. Это создает возможность его связывания с аналогичным доменом адапторной молекулы FADD (Fas-associated death domain), a затем – с таким же доменом белка RIP (receptor interacting protein). Образующийся DISC-комплекс (death-inducing signaling complex) активирует фермент – каспазу 8, последняя - эффекторные каспазы, вызывающие расщепление молекул-мишеней и развитие апоптоза. Исходя из рис. 12-3, можно заключить, что аналогичные события происходят при действии TNF (ФНО) через рецептор TNFR1. Только в этом случае с рецептором взаимодействует адапторный белок TRADD (TNFR-associated death domain), который передает сигнал апоптоза через другие белки (FADD и RIP). Выделяют и многие другие сигналы, ответственные за развитие апоптоза (протеинкиназы, протеинфосфатазы, онкосупрессоры р53 и р21, транскрипционные факторы c-mys, c-jun, c-fos, nur77 и т.д.).  Рис. 12-3. Молекулярные взаимодействия при индукции апоптоза через Fas и TNF-рецепторы. Обозначения: FasL и TNF — индукторы апоптоза; TNFR1 и Fas — рецепторы сигналов апоптоза; TRADD, FADD, RIP – белки, передающие сигнал апоптоза. Схематично сигнализация и последовательность внутриклеточных событий при развитии апоптоза, индуцированного внеклеточными и внутриклеточными сигналами, представлены на рис. 12-4. Данная схема иллюстрирует множественность пусковых воздействий и механизмов и единство конечных механизмов реализации апоптоза. В частности, доказано, что апоптоз может быть индуцирован многими внутриклеточными регуляторными факторами (LCK, ZAP-70, Ca2+, кальциневрин, NFAT, TRADD, FADD, RIP и др.), изменяющими активность каспаз цитоплазмы. Последние через влияние на свои ядерные мишени активируют эндонуклеазы, приводящие к деградации ДНК, и, таким образом, ответственны за универсальный этап индукции апоптоза. 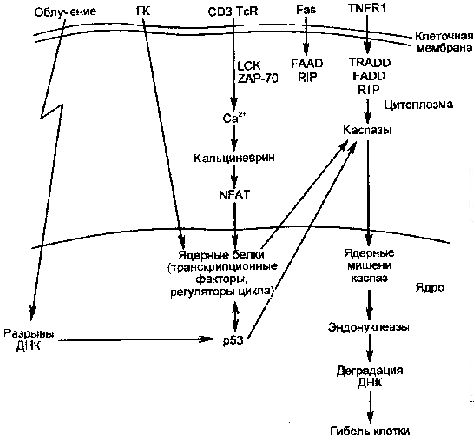 Рис. 12-4. Сигнализация и последовательность внутриклеточных событий при развитии апоптоза, индуцированного различными воздействиями Роль макрофагов в распознавании и удалении апоптотирующих клеток Различные апоптотирующие клетки, в том числе апоптотирующие гранулоциты и лимфоциты, быстро распознаются и поглощаются макрофагами. Выделяют три основных механизма распознавания и удаления клеток, подвергающихся апоптозу. Во-первых, макрофаги с помощью лектинов адгезируют различные клетки, вступающие на путь апоптоза. Это происходит в результате специфической перестройки углеводных компонентов мембран апоптотирующих клеток с потерей терминальных сиаловых кислот мембранными гликопротеидами, что приводит к экспрессии сахаров и снижению общего отрицательного потенциала наружной поверхности мембраны. Во-вторых, распознавание апоптотирующих клеток осуществляется с помощью макрофагального 43-интегринового рецептора, а также синтезируемого и секретируемого в микроокружение макрофагами тромбоспондина, выполняющего роль молекулярной «скрепки» между апоптотирующей клеткой и макрофагом. В-третьих, в распознавании апоптотирующих клеток участвует макрофагальный фосфатидилсериновый рецептор. Уже на ранних этапах программированной гибели в апоптотирующей клетке происходит инверсия мембранных фосфолипидов. Нейтральные фосфолипиды с наружного слоя перемещаются на внутренний, а отрицательно заряженные фосфолипиды с внутреннего слоя мембраны перемещаются на наружный слой. В результате экспрессии фосфатидилсерина наружу он распознается специфическим макрофагальным фосфатидилсериновым рецептором. Схематически эти основные механизмы распознавания апоптотических клеток макрофагами, представлены на рис. 12-5. 12.4. РОЛЬ АПОПТОЗА В ПАТОЛОГИИ За последнее десятилетие число опубликованных работ по проблеме апоптоза увеличилось более чем в 10 раз. Особенно возросло число исследований, посвященных роли апоптоза в развитии многообразной патологии, встречающейся в процессе жизни современного человека.  Рис. 12-5. Схема основных механизмов распознавания апоптотических клеток макрофагальными элементами В зависимости от вида и степени выраженности патологии клетка погибает, как отмечено выше, либо в результате апоптоза, либо некроза. В конечном итоге выбор организмом гибели клеток (апоптоз или некроз) зависит от содержания в этих клетках:
Доказано, что значительное снижение в клетках концентрации НАД+ и АТФ, повышение уровня активных форм кислорода (особенно Н2О2, О2–), NO и уменьшение количества ферментных и неферментных антиоксидантов, а также существенное повышение содержания провоспалительных цитокинов и белков острой фазы приводит к развитию и усилению некроза и одновременно к угнетению апоптоза. В последнее время особую роль в развитии как физиологических (в том числе апоптоза), так и патологических (в том числе некроза) процессов отводят важнейшему внутриклеточному медиатору – NO. NO может выполнять роль как молекулы-адаптогена, так и молекулы-разрушителя. Общеизвестно, что в малых (физиологических) количествах NO, определяемых активностью, главным образом, конститутивной нитрооксидсинтетазы, вызывает физиологические (апоптотические) изменения: активирует гуанилатциклазу, повышает содержание и действие цГМФ, снижает агрегацию тромбоцитов и адгезию нейтрофилов к эндотелию, обеспечивает внутриклеточный кальциевый гомеостаз, а также микробицидный и тумороцидный эффект фагоцитов. В больших количествах (на два порядка превышающих нормальные значения и достигающих сотен микромолей на 1 кг массы тканей) NO вызывает развитие дезадаптивных и деструктивных изменений, в частности, повышение проницаемости стенок сосудов, формирование и увеличение отека в тканях, развитие вазодилатации, кардиотоксическое действие, развитие острой сердечной недостаточности, в итоге проводящие к прогрессирующей, вплоть до необратимой, артериальной гипотензии. Известно, что резко сниженный уровень NO сопровождается снижением адаптации организма, а резко повышенный его уровень запускает суицидальную программу (самоуничтожение) клеточно-тканевых структур. И то, и другое необходимо корригировать, используя для этого различные пути, способы и средства лечения. Показано, что в условиях различной (и экзогенного, и эндогенного происхождения) патологии неизбежно возникают нарушения тех или иных звеньев как индукции, так и регуляции процесса апоптоза. Нарушение апоптоза участвует в качестве обязательного компонента формирования многих патологических процессов, при этом характер и выраженность развития патологического процесса определяется направленностью изменения апоптоза (ослаблением или усилением). 12.4.1. Апоптоз как обязательный компонент развития типовых патологических процессов Ведущая роль апоптоза в реализации многообразных физиологических процессов, в поддержании оптимального клеточного и тканевого гомеостаза, в обеспечении нормального развития клеточно-тканевых структур организма общеизвестна и не вызывает сомнений. Значение же апоптоза в формировании типовых патологических процессов может быть разным: решающим, незначительным и отсутствующим. В обобщающем варианте роль апоптоза в развитии различной патологии представлена на схеме 12-1. Как видно из схемы 12-1, апоптоз играет решающую роль в механизмах стресса, особенно в специфических стрессорных реакциях организма на действие различных интенсивных раздражителей. Общеизвестно, что типичным проявлением системного стресса являются лимфоцитопения и опустошение лимфоидных тканей и органов (тимуса, миндалин, лимфоузлов, пейеровых бляшек кишечника и др.), обусловленные действием избыточно образующихся при стрессорных реакциях глюкокортикоидных гормонов (особенно кортизола). Происходит гибель не только лимфоцитов, но и энтероцитов. Системная апоптотическая гибель клеток отмечается также при септическом шоке. В механизмах данного системного апоптоза важное значение имеют гиперпродукция макрофагами и другими клетками -ФНО (TNF-) и активация рецепторов TNFR1. При различных инфекционных процессах апоптотическая гибель клеток зависит, главным образом, от выделяемых микроорганизмами экзотоксинов. Последние в виде суперантигенов выступают в качестве индукторов активационного апоптоза. Сам же инфекционный типовой патологический процесс не является обязательным в развитии апоптоза. Апоптотическое повреждение клеток, возникающее под действием Т-киллеров, отмечается при различных аутоиммунных процессах. Локальная апоптотическая гибель клеток обнаруживается также при постишемической реоксигенации. На начальных этапах развития как лихорадки, так и воспаления апоптоз не является обязательным компонентом в механизмах их развития. Ибо в начале воспалительного процесса гибель клеточно-тканевых структур в очаге воспаления развивается благодаря не апоптозу, а некрозу, сопровождающемуся выходом внутриклеточных структур (в том числе активных гидролаз) наружу, что приводит к последующей некротической 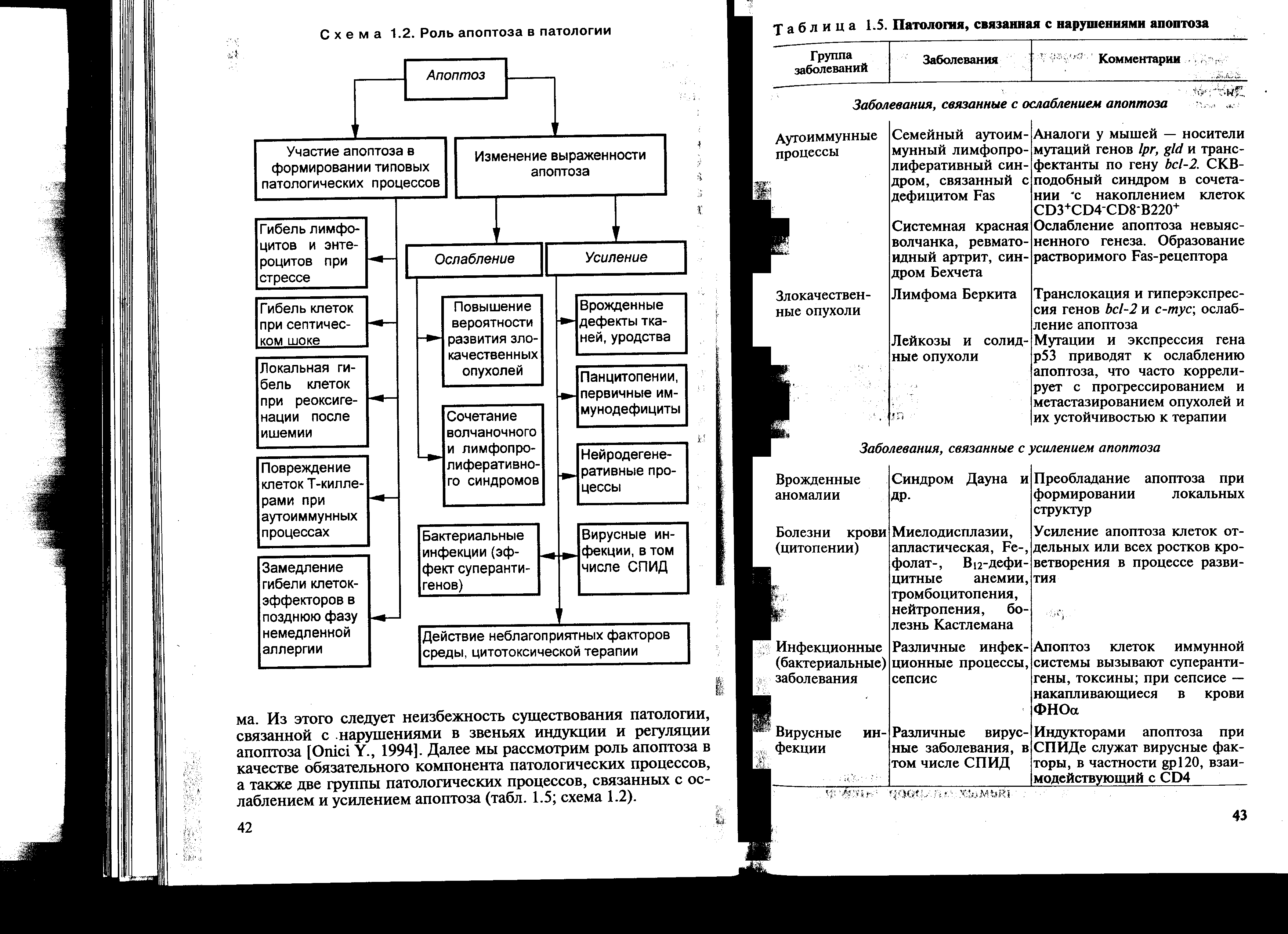 Схема 12-1. Роль апоптоза в патологии гибели соседних клеточных и внеклеточных образований, вплоть до их гнойного расплавления. По мере протекания воспаления, особенно на поздних этапах его развития, процесс апоптоза начинает занимать все большее и большее место в гибели выполнивших свои защитные функции иммунных клеток (в том числе фагоцитов) с целью более быстрого их обновления. При аллергическом воспалении, особенно в позднюю фазу ГНТ, отмечается замедление элиминации, а значит затруднение гибели эффекторных клеток. Это обусловлено способностью данных клеток к самоподдержанию благодаря выработке аутокринных цитокинов (например, ГМ-КСФ), защищающих эти клетки от апоптоза. Известно, что в развитии опухолей и реализации их отрицательного действия на целостный организм участие апоптоза не является обязательным. В то же время доказано, что при угнетении апоптоза значительно замедляется элиминация как генетически измененных собственных клеток, так и поступивших в организм чужеродных клеток. Таким образом, апоптоз может как быть (что реже), так и не быть (что чаще) обязательным компонентом различных видов патологий (патологических процессов и заболеваний). 12.4.2. Патология, связанная с изменением выраженности апоптоза В настоящее время доказанно, что в развитии различных видов патологии могут иметь значение как ослабление процесса апоптоза, так и его усиление. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||