патологическая физиология. Учебное пособие для самоподготовки студентов медицинских факультетов к разделу курса патофизиологии Патофизиология обмена веществ пособие составлено
 Скачать 277 Kb. Скачать 277 Kb.
|

МукополисахаридозыМукополисахаридозы - нарушение расщепления гликозаминогликанов (ГАГ). ГАГ, или кислые гетерополисахариды, являются полимерами и состоят из следующих мономеров: глюкуроновая кислота, сульфаты, гексозамины, гексозы. В состав ГАГ входят также белки. Под действием гликозидаз ГАГ расщепляются на свои компоненты. Сульфаты выводятся из организма, а остальные идут на синтезы других веществ. При дефектах ферментов ГАГ не расщепляются, а накапливаются в лизосомах, выводятся с мочой. Для большинства мукополисахаридозов характерно тяжелое поражение опорно-двигательного аппарата, часто карликовый рост больных, деформированные конечности, гротескные гримасы, застывшие на лице. Имеются висцеромегалия, большой живот, характерны грубая кожа, избыточное оволосение, часто наблюдаются болезни сердца, легких, при многих мукополисахаридозах развивается умственная отсталость, поражаются органы чувств. Основные показатели обмена углеводов и их нарушения. I. Содержание глюкозы в крови и моче. Содержание глюкозы в крови в норме составляет 3,3-6,1 ммоль/л. Источниками ее являются: углеводы пищи (переваривание, унификация) гликоген печени (гликогенолиз), глюконеогенез. Глюкоза, находящаяся в крови, в дальнейшем поступает: в ткани, где активизируется, превращаясь в глюкозо-6-Ф, и участвует в различных процессах. в почки, где подвергается фильтрации, а в последующем почти вся реабсорбируется, т.е. в норме глюкозы в моче почти нет. В лабораторной практике широко используется определение содержания глюкозы в крови и моче. Содержание глюкозы в крови может повышаться – гипергликемия либо понижаться – гипогликемия. Гипогликемия – снижение уровня сахара в крови < 2,8 ммоль/л.
У новорожденных причинами гипогликемий могут быть: внутриутробная гипотрофия, плацентарная недостаточность – неадекватное поступление питательных веществ в организм ребенка гипоксия, гипертермия, гипотермия, синдром дыхательных расстройств, инфекции – интенсивное поглощение углеводов наследственные аномалии обмена углеводов (галактоземия, непереносимость фруктозы и др.) гиперинсулинизм детей, рожденных от матерей с сахарным диабетом недостаточность коры надпочечников Частота гипогликемии возрастает при низкой массе тела. Гипергликемия – увеличение содержания сахара в крови > 6,1 ммоль/л.
Если содержание глюкозы в крови превышает 9 ммоль/л (т.н. почечный порог реабсорбции), то глюкоза появляется в моче – глюкозурия. Возможна глюкозурия без гипергликемии при блокаде ферментов реабсорбции глюкозы в почечных канальцах (почечная форма глюкозурии). II. Толерантность организма к углеводам. Толерантность - устойчивость организма к углеводам (переносимость). Толерантность организма к углеводам определяют с помощью сахарной нагрузки. В настоящее время этот метод используют в диагностических целях только беременным женщинам и в сложных диагностических ситуациях. Сущность метода заключается в том, что у человека натощак берут первую порцию крови, затем дают выпить раствор глюкозы или сахара (1 г на 1 кг веса) и через каждые 30 мин., в течение 2-ух часов, берут следующие порции крови, определяют во всех пробах уровень содержания глюкозы и строят сахарные кривые: 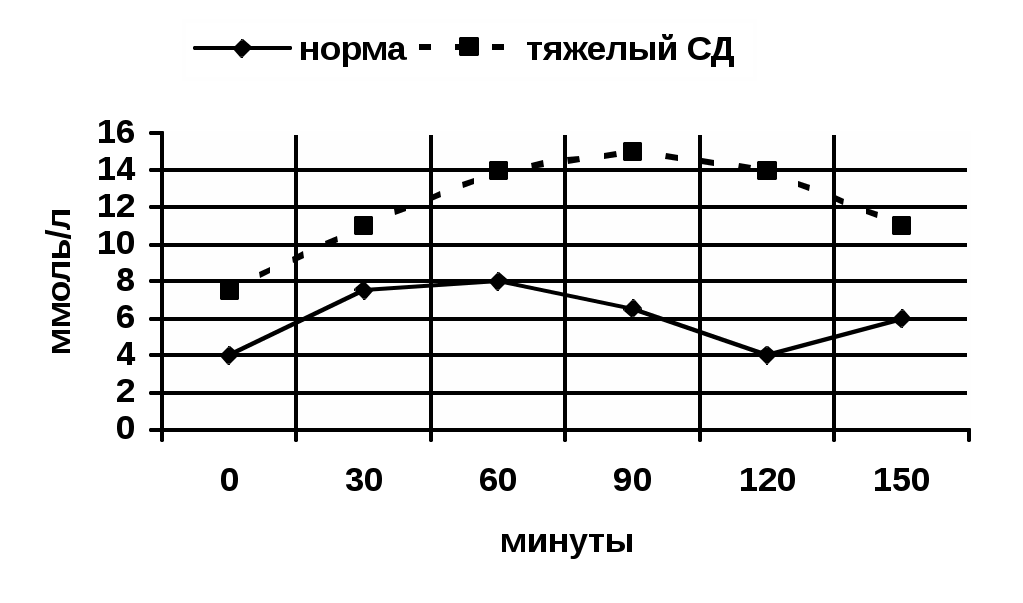 Также определяют коэффициент Бодуэна (кБ). Б - А кБ = _______ х 100% А А - исходная концентрация глюкозы. Б - макс. прирост уровня глюкозы. В норме кБ = 50-75%. В условиях патологии толерантность может быть повышена либо понижена. Толерантность повышена: При избытке инсулина (инсулома). При недостатке контринсулиновых гормонов (глюкагон, адреналин, глюкокортикостероиды, тироксин). Характеризуется тем, что при сахарной нагрузке происходит незначительное повышение глюкозы в крови и быстрая его нормализация. Толерантность понижена: -При сахарном диабете -При избытке контринсулиновых гормонов. Характеризуется высоким приростом глюкозы в крови при сахарной нагрузке и медленной нормализацией. Патофизиология белкового обмена. Функции белков. Понятие об азотистом балансе. Белки - основная и необходимая составная часть всех живых организмов. Они выполняют целый ряд разнообразных функций: ферментативную; регуляторную; структурную; сократительную; иммунную; транспортную опорную; энергетическую. В пересчете на сухой вес белки составляют 44% массы тела. Ежедневное рекомендуемое потребление белка взрослыми 1-1,5 г в день на кг веса. Для лиц, занятых тяжелым физическим трудом, потребление белка необходимо увеличить до 2 г/кг. Грудным детям и в период полового созревания необходимы 1,5-2 г/кг белка в сутки. Потребность в белке повышается при лактации, беременности, регенерации, стрессе, акклиматизации и др. Нарушения белкового обмена являются компонентами патогенеза всех патологических процессов. Показателем белкового обмена служит азотистый баланс - понятие, характеризующее соотношение процессов анаболизма и катаболизма. У взрослого здорового человека количество выводимых из организма азотистых веществ равно тому, которое поступает в организм с пищей, то есть азотистый баланс нулевой (азотистое равновесие). Положительный азотистый баланс или преобладание процессов анаболизма над катаболизмом наблюдается в случаях, когда количество азота, поступающее в организм, превышает его выведение. Это наблюдается в растущем организме, при беременности, увеличении продукции СТГ, половых гормонов, инсулина, после голодания, в период реконвалесценции. Отрицательный азотистый баланс характеризуется преобладанием процессов распада белков, т.е. азотистых соединений из организма выводится больше, чем поступает с пищей. Отрицательный азотистый баланс наблюдается при голодании, неполноценном белковом питании, инфекциях, интоксикациях, повышенных потерях белка (через кожу, почки, кишечник), тиреотоксикозе, сахарном диабете, усиленном синтезе глюкокортикоидов, стрессовых ситуациях. Результатом отрицательного азотистого баланса являются дистрофия и нарушения иммунитета. В процессе обмена белков в организме различают несколько этапов: усвоение пищевых белков и всасывание аминокислот в ЖКТ и последующий их метаболизм в клетках и тканях; межуточный обмен аминокислот; конечный этап белкового обмена. Патология обмена белков возможна на любом из этапов их метаболизма. I Нарушение усвоения пищевых белков и всасывания аминокислот в ЖКТ. Усвоение белков пищи и их превращения в организме – сложный процесс, он осуществляется в желудке, тонком кишечнике и в клетках слизистой тонкого кишечника. Расщепление белков происходит неравномерно. В желудке под влиянием пепсина отщепляются тирозин и триптофан, другие же аминокислоты отщепляются в кишечнике. Трипсин и химотрипсин расщепляют белки до олигопептидов, которые затем всасываются слизистой тонкого кишечника, где расщепляются до аминокислот под влиянием внутриклеточных пептидаз и вместе с ионами Nа+ диффундируют в кровь воротной вены. Карбоксипептидазы расщепляют белки до аминокислот, которые быстро проникают в слизистую тонкого кишечника и в кровь. Этиопатогенез нарушения усвоения белков пищи и всасывания аминокислот: голодание – недостаток белка в пище и, как следствие, дефицит ферментов гипертермия, лихорадка, интоксикации, инфекции, авитаминозы, заболевания желудка (воспалительные процессы, язвенная болезнь, опухоли), патология поджелудочной железы (воспалительные процессы, нарушение кровообращения, травмы, опухоли, резекции), т.е. ряд заболеваний, приводящих к дефициту пищеварительных ферментов усиление перистальтики кишечника, ведущее к снижению времени действия фермента на субстрат воспаление, резекции, опухоли кишечника и, как следствие, недостаточная поверхность для всасывания медиаторы лихорадки и стресса могут нарушать мембранное пищеварение Последствиями нарушения усвоения белков являются: отрицательный азотистый баланс; снижение массы тела; ослабление иммунитета анемия; отеки; нарушение транспорта веществ кровью; гипопротеинемия; аллергизация – в случае поступления в кровоток нерасщепленных до аминокислот олигопептидов. Поскольку в организме нет депо белков, то поступающие с пищей белки должны восполнять их расход и потребность в незаменимых аминокислотах. Недостаточное поступление белка в организм приводит к белково-калорийной недостаточности, которая в основном проявляется в виде квашиоркора и алиментарной дистрофии. Квашиоркор (“болезнь отнятых от груди") - несбалансированная алиментарная недостаточность белка в детском возрасте. Это заболевание сопровождается целым рядом тяжелых симптомов: отрицательный азотистый баланс; снижение массы тела; гипопротеинемия; отеки, асцит; комбинированные ИДС; задержка физического и умственного развития; выраженные изменения кожи (т.н. "красный мальчик"); поражения печени, поджелудочной железы; анемия. Прогноз при квашиоркоре неблагоприятный, большой процент летальных исходов. Алиментарная дистрофия или алиментарный маразм - сбалансированная белково--калорийная недостаточность, приводит к мобилизации белка из костей, мышц, кожи, при тяжелых состояниях - из внутренних органов и в последнюю очередь из мозга . Проявления: отрицательный азотистый баланс; снижение массы тела; гипогликемия; гиперкортицизм; повышенный уровень глюкагона и соматостатина; гипопротеинемия; кетонемия; клеточный иммунодефицит; задержка физического и умственного развития. При алиментарном маразме нет резко выраженной гипопротеинемии, отеков, расстройства обмена электролитов, отсутствуют специфические изменения кожи. Исхудание, как правило, начинается с лица, придавая ему старческий вид, у детей наблюдается "обезьянье лицо". Прогноз в данном случае благоприятен при своевременно начатом и правильном лечении. Отрицательно сказывается и избыток белка, поступающего в организм с пищей. Это приводит к положительному азотистому балансу, диспепсии, дисбактериозу, кишечной аутоинфекции и аутоинтоксикации. Качественное белковое голодание. Белки пищи содержат 22 аминокислоты, из них 8 - незаменимых. Нарушение соотношения аминокислот в белке, потребляемом с пищей, приводит к появлению общих симптомов: отрицательный азотистый баланс, замедление роста и нарушение развития у детей; нарушение процессов регенерации тканей, снижение массы тела и аппетита. Относительный дефицит даже одной аминокислоты может приводить к затруднению процесса синтеза белков, создавать избыток других аминокислот с накоплением в организме промежуточных продуктов обмена этих аминокислот. Так, дефицит незаменимых аминокислот вызывает специфические проявления: недостаток гистидина ведет к дерматитам, анемиям, снижению продукции гистамина, ухудшению умственной деятельности; изолейцина и лейцина – к поражению почек, щитовидной железы; лизина – анемиям, остеопорозу, поражению печени, легких; головной боли и повышенной чувствительности к шуму; метионина – к патологии печени, почек, надпочечников, облысению и ускорению атеросклероза; фенилаланина и тирозина – к патологии щитовидной железы и надпочечников; аргинина – нарушению сперматогенеза; триптофана – к анемии и катаракте; валина - к нарушению координации движения; треонина – к отекам и падению веса. Недостаток любой из аминокислот способен замедлить рост у детей. Избыток отдельных аминокислот также приводит к разнообразным нарушениям. Общие проявления: нарушение вкуса; снижение аппетита; уменьшение массы тела; нарушения функций органов и тканей; расстройства обмена других аминокислот (избыток лейцина подавляет обмен валина), избыток тирозина ведет к гиперкатехоламинемии и гиперкортицизму; фенилаланина – задержке психомоторного развития ребенка, слабоумию, экземам; метионина – к гемолитической анемии, сердечной и печеночной недостаточности). Нарушение синтеза и распада белков в тканях (конформационные изменения белков). Для нормального синтеза белков необходимо правильное и активное функционирование соответствующих генетических структур, на которых совершается этот синтез. Повреждение генетического аппарата может быть приобретенным и наследственным Результатом этого является измененный синтез белков или синтез белков с измененной структурой. Последнее может проявиться нарушением аминокислотного состава белковой молекулы, (например, молекула гемоглобина при серповидно-клеточной анемии), укорочением молекул (когда транскрипция информации с ДНК-матрицы идет только до дефекта в ней; эти полипептиды обладают низкой биологической активностью), а также синтезом аномально длинных белков, если мутация произошла в «стоп-сигнале» гена и терминирующий кодон исчез. Примером этого может служить появление удлиненных α-цепей гемоглобина; нерегулируемая скорость синтеза матричной РНК при нарушении функционирования гена-регулятора или оператора, синтез соответствующих белков независимо от потребности в них. Измененный синтез белков может быть следствием нарушения одного из звеньев белоксинтезирующей системы - аппарата трансляции либо посттрансляционной модификации молекул. С увеличением частоты ошибок трансляции в процессе жизни связывают старение организма. По-видимому, относительная нехватка транспортной РНК может ограничивать функциональные возможности органов и тканей при повышенных нагрузках (адаптация, регенерация, стресс). Одной из причин нарушения считывания генетического кода являются структурные изменения в рибосомах, которые могут возникать, например, при введении стрептомицина. Нарушение синтеза белков может быть связано с дефектом регуляции, на клеточном уровне - это воздействие метаболитов, на уровне органов и организма – гормонов и нервной системы. Известно, что при повышенной продукции соматотропина увеличивается синтез белков, наблюдается усиленный рост молодого организма, ведущий к гигантизму. Недостаточность этого гормона выбывает противоположный эффект. В денервированных тканях при перерезке нервов не только снижается выработка белков, выражающаяся развитием атрофии; но и появляются качественно новые белки – аутоантигены. В то же время реинннервация (или первичная иннервация в онтогенезе) индуцирует радикальную перестройку метаболизма, в том числе биосинтеза белков. Это наглядно показано в экспериментах с перекрестной реиннервацией, когда врастающий в мышцу «чужой» нерв сообщает ей новые свойства, а также индуцирует синтез белков, не свойственных мышце. Одним из проявлений нарушений распада и синтеза белков в тканях являются диспротеинемии. Диспротеинемии. Кровь здорового человека содержит 5 белковых фракций: альбумины, α1- глобулины, α2-глобулины, β-глобулины, γ-глобулины. Нормальное количество и соотношение между фракциями белков плазмы обозначается как эупротеинемия. Изменения в качественном и количественном соотношении белков наблюдаются почти при всех патологических состояниях. Нарушения соотношения между разными фракциями белков крови называют диспротеинемиями, к ним относят гиперпротеинемию – увеличение концентрации белков плазмы, гипопротеинемию – уменьшение концентрации белков в плазме крови. Если изменения касаются только глобулиновых фракций, говорят о дисглобулинемии. Гиперпротеинемия может быть ложной или относительной, что наблюдается при сгущении крови (дегидратация), и истинной. Истинная гиперпротеинемия наблюдается только при парапротеинемии и, практически всегда, касается глобулиновых фракций, т.е по сути она является гиперглобулинемией. При усиленном синтезе иммуноглобулинов количество белка, может достигать 150-160 г/л (в N 60-78 г/л), что встречается при миеломной болезни, макроглобулинемии Вальденстрема, действии поликлональных иммуностимуляторов. Парапротеинемии – появление в крови патологических протеинов, образующихся в результате пролиферации мутантных линий плазматических клеток. Наиболее часто (15% онкогематологических заболеваний) встречается миеломная болезнь (болезнь Рустицкого-Калера), при которой в больших количествах вырабатываются IgG, IgM, IgD, IgA, IgE плазматическими клетками опухоли, это связано с экспрессией и транслокацией онкогена в 14 хромосому, где он локализуется по соседству с генами иммуноглобулинов. Второе место занимает макроглобулинемия Вальденстрема или лимфоцитарная лимфома, при которой в большом количестве неопластические клетки вырабатывают IgM. Это заболевание сопровождается тромбоцитопенией, анемией, повышенной вязкостью крови. При всех парапротеинемиях в моче обнаруживают легкие цепи иммуноглобулинов - белок Бенс-Джонса. Разновидностью парапротеинов являются криоглобулины - патологические белки со свойствами глобулинов, которые образуются при охлаждении и свойственны таким заболеваниям, как миеломы и лимфомы. Появление в крови криоглобулинов вызывает повреждение сосудистой стенки, образование тромбов и осложняет патологический процесс. Криоглобулином является также фибронектин, присутствующий в плазме некоторых людей в аномально больших количествах. Гипопротеинемии также различают ложные (псевдогипопротеинемии), которые сопровождают выраженную гемодилюцию, и истинные. В свою очередь, истинные гипопротеинемии делят на первичные (наследственные) и вторичные (приобретенные). Первичные гипоальбуминемии характерны для недоношенных детей с их незрелой печенью. Среди них выделяют анальбуминемию – аутосомно-рецессивное, крайне редко встречающееся заболевание, при котором практически полностью отсутствуют альбумины (3%). Компенсаторно увеличивается синтез глобулинов. Анальбуминемия сопровождается гиперальдостеронизмом, отеками, гиперхолестеринемией, низким ростом и судорожными припадками. Врожденная агаммаглобулинемия наблюдается при иммунодефицитах, в частности при сцепленном с Х-хромосомой рецессивном синдроме Брутона. При этом количество глобулинов падает до 1 г/л. Одновременное снижение и глобулинов, и альбуминов отмечается при экссудативной энтеропатии у детей, которые страдают хронической неинфекционной диареей. Вторичные гипопротеинемии встречаются значительно чаще и представляют собой, как правило, гипоальбуминемию на фоне гиперглобулинемии.Это наблюдается при: пищевой белковой недостаточности нарушении поступления аминокислот из кишечника (энтериты, инфекционные заболевания, наследственные дефекты переваривания и всасывания белков, панкреатическая недостаточность); печеночной недостаточности (гепатиты, циррозы); усиленных внекишечных потерях белка (через почки , кожу,путем перераспределения белка между кровью и тканями при шоке, васкулитах, действии БАВ); синдроме Иценко – Кушинга вследствие усиленного катаболизма альбумина плазмы. II Нарушение обмена аминокислот в тканях. Аминокислоты, образующиеся в результате расщепления белков, поступают в кровь. Кроме того, они образуются при деструкции тканевых белков под действием внутриклеточных катепсинов (протеиназ). Основная часть аминокислот используется в организме в качестве строительных блоков при синтезе белков, а так же для синтеза пуриновых и пиримидиновых оснований, гормонов, гема, различных биологически активных пептидов (интерлейкины, факторы роста и т. д.), меланина, глюкозы, жирных кислот и ряда других веществ. Глицин и глутамат играют роль нейромедиаторов в ЦНС. Аминокислоты, не использованные для вышеупомянутых целей, подвергаются окислению до СО2 и Н2О с освобождением энергии (в норме при окислении аминокислот освобождается 10-15% образующейся в организме энергии). Обмен аминокислот усиливается при избыточном поступлении их в организм, при голодании, сахарном диабете, гипертиреозе, снижении синтеза белков и некоторых других состояниях. В процессе метаболизма аминокислоты подвергаются реакциям трансаминирования, дезаминирования и декарбоксилирования. Причем трансаминирование направлено на образование аминокислот, а декарбоксилирование – на их разрушение. Сущность трансаминирования заключается в переносе аминогруппы с аминокислоты на кетокислоту (чаще – на α-кетоглутарат или оксалоацетат). В результате образуются аминокислота (из α-кетоглутарата – глутаминовая) и та или иная кетокислота (например, из аланина - пировиноградная). Практически все аминокислоты подвергаются реакции трансаминирования, кроме треонина. Процесс трансаминирования катализируется трансаминазами, коферментом которых является пиридоксальфосфат (В6). Образовавшаяся при реакции трансаминирования глутаминовая кислота подвергается окислительному дезаминированию, т.е. отщеплению аминогруппы под действием глутаматдегидрогеназы с образованием иона аммония и α-кетоглутаровой кислоты, которая может снова вступить в реакцию трансаминирования или окислиться в цикле трикарбоновых кислот. Кетокислоты, образующиеся при трансаминировании, (например, пировиноградная), также могут окислиться до СО2 и Н2О подобно глюкозе и жирным кислотам. Поскольку реакции трансаминирования и окислительного дезаминирования могут идти как в прямом, так и в обратном направлении, то они играют роль не только в превращении аминокислот в кетокислоты, но и в образовании из кетокислот ряда заменимых аминокислот в том случае, если организм испытывает в них потребность. Кроме того, кетокислоты могут быть использованы для синтеза глюкозы. Нарушение трансаминирования в целом организме происходит при гиповитаминозе В6 (голодание, сахарный диабет), а в отдельных органах, например, в печени – при некрозе клеток, что сопровождается выходом трансаминаз в кровь. Такое же явление имеет место при инфаркте миокарда. В поврежденных клетках может быть нарушен синтез белковой части трансаминаз. Процесс окислительного дезаминирования снижается не только в связи с ослаблением трансаминирования, но и при гипоксии, гиповитаминозах В2, РР, С, белковом голодании. Нарушение процессов трансаминирования и окислительного дезаминирования аминокислот ограничивает их использование для синтеза глюкозы, жирных кислот, заменимых аминокислот, а также их окисление с освобождением энергии. При этом повышается содержание свободных аминокислот в сыворотке крови (гипераминоацидемия). Гипераминоацидемия приводит к различного рода патологическим состояниям. Так избыток метионина ведет к анемиям, некрозу печени, миокардиодистрофии, отставанию в росте в детском возрасте; гистидина – к задержке умственного развития. Избыточное содержание аминокислот в крови ведет к гипераминоацидурии, снижению синтеза мочевины. Такие нарушения особенно выражены при обширных повреждениях гепатоцитов (вирусные и токсические гепатиты и др.), так как в этих клетках метаболизм аминокислот происходит наиболее интенсивно. Наряду с вышеупомянутой внепочечной гипераминоацидурией, обусловленной усиленным поступлением аминокислот из крови в мочу, существует почечная форма гипераминоацидурии, связанная с нарушением реабсорбции аминокислот в почечных канальцах. При этом содержание аминокислот в сыворотке крови нормально или даже понижено. Гипераминоацидурия (физиологическая) может наблюдаться у детей раннего возраста в связи с функциональной неполноценностью (незрелостью) эпителия почечных канальцев; у беременных женщин повышается экскреция с мочой гистидина и ряда других аминокислот. Одним из путей метаболизма аминокислот является декарбоксилирование. Оно состоит в отщеплении от аминокислоты карбоксильной группы с образованием СО2 и биогенных аминов. Этой реакции подвергаются только некоторые из аминокислот, например: гистидин – с образованием гистамина; тирозин – тирамина; глутаминовая кислота – аминомасляной кислоты; 5-гидрокситриптофан – серотонина и др. Биогенные амины обладают высокой фармакологической активностью, изменение их количества приводит к целому ряду патологических явлений в организме. Декарбоксилирование катализируется декарбоксилазами, коферментом которых является пиридоксальфосфат (витамин В6), при его дефиците образование биогенных аминов снижается. В частности, уменьшается образование аминомасляной кислоты, которая является основным тормозным нейромедиатором, как следствие этого наблюдается частое развитие судорог. Серотонин и дофамин являются также нейромедиаторами ЦНС, их повышенное или пониженное содержание в ткани мозга играет роль в патогенезе некоторых форм нейропатологии (нервная депрессия, паркинсонизм, шизофрения). Повышенное образование в организме серотонина, наиболее выраженное при опухолях из энтерохромафинных клеток кишечника, сопровождается спазмом мускулатуры бронхов и кишечника, диареей, усилением агрегации тромбоцитов; кроме того, серотонин является мощным вазоконстриктором. Хорошо известна роль гистамина в появлении болевых ощущений, развитии воспаления и аллергических реакций, в том числе анафилактического шока. Устранение избытка биогенных аминов происходит при участии аминооксидаз, которые катализируют превращение их в альдегиды после отщепления аминогруппы в виде NН3. Серотонин превращается в оксииндолилуксусную кислоту, которая выделяется с мочой. Наследственные нарушения обмена аминокислот. Прохождение аминокислот через определенные метаболические пути детерминируется наличием и активностью соответствующих ферментов. Наследственное нарушение синтеза ферментов приводит к тому, что нарушается метаболизм аминокислот, они накапливаются в организме и появляются в биологических средах: моче, поте, кале, спинномозговой жидкости. Клиническая картина таких заболеваний определяется, во-первых, появлением слишком большого количества веществ, которое должно было метаболизироваться при участии заблокированного фермента, во-вторых, дефицитом вещества, которое должно было образоваться. К таким нарушениям относятся фенилкетонурия, алкаптонурия, альбинизм. Фенилкетонурия – заболевание передается рецессивным путем, связано с дефицитом фермента фенилаланингидроксилазы, что приводит к нарушению превращения фенилаланина в тирозин. Фенилаланин в больших количествах (в 50-100 раз больше нормы, норма-0,015 г/л) накапливается в крови, тканях, цереброспинальной жидкости, выводится с мочой. Проявления: фенилкетонурия; олигофрения; недостаточное образование адреналина и норадреналина (избыток фенилаланина тормозит активность фермента – дофамингидроксилазы, необходимого для синтеза катехоламинов). Диагностический тест: добавление к моче трихлоруксусного железа дает зеленое окрашивание. Алкаптонурия – передается рецессивным путем и связана с дефицитом фермента оксидазы гомогентизиновой кислоты, что нарушает процесс превращения гомогентизиновой кислоты в малеилацетоуксусную, и гомогентизиновая кислота накапливается и с кровью попадает в ткани – хрящи, сухожилия, связки, внутренний слой стенки аорты. Проявления: темные пятна в области ушей, носа, щек, на склерах; охроноз (темное окрашивание в соединительной ткани и хрящах); поражения сердечно – сосудистой системы; артриты; Диагностический тест – моча на свету чернеет. Альбинизм – при отсутствии фермента тирозиназы не синтезируется красящий пигмент - меланин. Проявления могут быть местного характера (обесцвеченная прядь волос) и общего: кожа молочно – белого цвета; обесцвеченные волосы, радужка глаз; светобоязнь, снижение остроты зрения; глухота, немота; эпилепсия; полидактилия; изредка олигофрения. III Нарушения конечных этапов белкового обмена, синтеза мочевины. Гиперазотемия. Конечные этапы белкового обмена – процессы образования и выведение из организма азотсодержащих продуктов (аммиак, мочевина, мочевая кислота, креатинин, индикан). Показателем, характеризующим состояние этого этапа белкового обмена, является содержание остаточного азота в крови, который распределяется следующим образом: азот мочевины азот аминокислот азот мочевой кислоты азот креатина и креатинина остальных азотистых продуктов Главная составная часть остаточного азота – мочевина (50%). Таким образом, нарушения конечных этапов обмена белков связаны с нарушением образования мочевины в печени или нарушением выделительной функции почек и проявляются они гиперазотемией в 3-х формах: печеночная или продукционная; почечная или ретенционная; смешанная, т.е. ретенционная + продукционная. Продукционная гиперазотемия. Причины: поражение печени (гепатит, цирроз, отравление ядами): значительный перекорм белками; гипоксия; голодание. Изменение остаточного азота при этой форме характеризуется повышением содержания немочевинных фракций остаточного азота, что связано с нарушением образования мочевины в печени. Нарастает содержание аммиака и аминокислот. Клинические проявления продукционной гиперазотемии: гепатогенная энцефалопатия (нарушение сна, эмоциональная лабильность, изменения на ЭЭГ, кома); Ретенционная гиперазотемия. Причины. острая и хроническая почечная недостаточность; воспаление; расстройство кровообращения почек; нарушение оттока мочи. Основной механизм этого типа гиперазотемии – недостаточное выделение азотистых продуктов с мочой вследствие нарушения почечных функций. Растет содержание остаточного азота и азота мочевины (в тяжелых случаях остаточный азот может повышаться до 200-300 мг/дл), что ведет к отравлению азотистыми продуктами, нарушению водно - электролитного обмена, КОС, уремической коме и летальному исходу. Быстрое повышение остаточного азота свойственно комбинированным нарушениям, когда страдают функции почек и печени. Такая форма, при которой сочетается повышенный распад белка в тканях и недостаточное выведение азотистых продуктов с мочой, может быть при шоке, гепато-ренальном синдроме, острой паренхиматозной желтухе, портальной гипертензии, синдроме раздавливания, неукротимой, рвоте, поносах. Повышенное содержание мочевой кислоты в крови (гиперурикемия) ведет к подагре, одному из основных заболеваний связанных с нарушением пуринового обмена. В N содержание мочевой кислоты 0,18- 0,48 ммоль/л. Подагра встречается в 2-18% случаев, относится к полигенным заболеванием. Возникновению подагры способствуют болезни почек, злоупотребление алкоголем, пивом, физическая активность, отравление солями тяжелых металлов. 95% больных подагрой – мужчины, так как аллели, предрасполагающие к гиперурикемии, находятся в Х – хромосоме (гены гипоксантинфосфорибозилтрансферазы – ГФРТ и фосфорибозилпирофосфатсинтетазы – ФРПФ), а эстрогены обладают антиурикемическим действием. Чаще подагра поражает лиц среднего и пожилого возраста. В основе патогенеза подагры лежит накопление уратов в хрящевой ткани и почках, а так же механизмы провоцируемого ими воспаления. Заболевание протекает приступообразно с поражением суставов (как правило, моноартрит), отложением кристаллов мочекислого натрия в тканях (подагрические шишки), нефропатией и мочекаменной болезнью. Другой формой нарушения пуринового обмена является гипоурикемия, вызванная низким содержанием мочевой кислоты в крови и чаще протекающая бессимптомно. Из заболеваний нарушения обмена пиримидиновых оснований встречается оротацидурия, связанная с дефектом ферментов оротатфосфорибозилтрансферазы и оротодин-5-осфатдекарбоксилазы белкового комплекса, кодируемого в длинном плече третьей хромосомы. У детей с таким дефектом, наследуемым аутосомно-рецессивно, замедляется рост, возникает мегалобластическая анемия, резистентная к витаминотерапии, лейкопения со сдвигом формулы нейтрофилов влево. Характерен иммунодефицит клеточного типа. Выведение оротовой кислоты с мочой ведет к закупорке мочеточников и уретры. Патофизиология липидного обмена |