Самохвалов В.П. - Психиатрия (копия). Учебное пособие для студентов медицинских вузов (2002 г.)
 Скачать 0.72 Mb. Скачать 0.72 Mb.
|

|
Эпилепсия с большими судорожными приступами Grand mal во время пробуждения Этиология Вероятно, это форма вытекает из нелеченных или недолеченных пикнолепсий детской и ювенильной. Эпилептическая система при этом изменяется, поэтому в каждом случае приходится ее уточнять и подбирать медикамент индивидуально. В связи с малой проявляемостью этих эпилепсии на ЭЭГ, целесообразно уточнение эпилептической системы нейропсихологическими методами. Генетическая предрасположенность довольно четкая; от 4 до 12% членов семьи страдают эпилептическими приступами. Распространенность Приблизительно 25% всех эпилепсии с «большими припадками» следует относить к данному синдрому. Клиника Начало — синдром чаще развивается на 2-м десятилетии жизни, преимущественно в период полового созревания. В подавляющем большинстве случаев припадки ГТКП (генерализованный тонико-клонический припадок) возникают вскоре после пробуждения (90%), независимо от времени пробуждения. Второй суточный пик припадков — в вечернее время, в релаксации. Существует выраженная корреляция с повышенной светочувствительностью. Приступы — обычный большой развернутый припадок сразу после пробуждения или в течение 1—1,5 часа после пробуждения. Если имеется при этом другой тип припадков, то это скорее всего абсансы или миоклонические припадки. Диагностика Основывается на типичности приступов и времени их возникновения. ЭЭГ уточнение проблематично, так как приступы скорее будут регистрироваться в «фазовых» состояниях, когда человек «не до конца проснулся». Помогает депривация сна. До 30% пациентов выявляют светочувствительность. Прогноз Даже при устойчивой терапевтической ремиссии припадков прекращение лечения следует начинать не ранее чем спустя пять лет после исчезновения приступов при наличии хороших результатов на ЭЭГ, а также после достижения двадцатилетнего возраста. Терапия Достаточный и регулярный сон, соблюдение режима сна-бодрствования имеет важное значение. На первом месте — вальпроаты, на втором — фенитоины. Применение карбамазепинов противопоказано, так как при этой форме нередки абсансы и миоклонические приступы. Ювенильная миоклоническая эпилепсия (эпилепсия с импульсивными Petit Mal, ЮМЭ, с миоклоническим Petit Mal, синдром Янца, синдром Герпина — Янца) Этиология Форма генерализованной идиопатической эпилепсии с выраженным генетическим предрасположением, идентифицированным генетическим дефектом (короткое плечо 6-й хромосомы на расстоянии 21 сМ от теломеры и локус 15ql4). Распространенность Синдром частый, составляет около трети случаев с дебютом в подростковом возрасте и до 11—12% среди всех форм эпилепсии. Клиника Возраст начала: 12—20 лет. Приступы короткие, «простреливающие», билатерально-синхронные, массивные, симметричные миоклонии, преимущественно в руках и верхнем плечевом поясе, в большинстве случаев с сохраненным сознанием. При вовлечении ног — внезапное падение. Иногда припадки следуют залпами. Как правило, после пробуждения при движении, провоцируются бессонницей. Обычно комбинируются с генерализованными тонико-клоническими приступами, которые возникают или при пробуждении, или вечером в состоянии расслабления («эпилепсия конца рабочего дня»). Неврология без особенностей, иногда — фокальная микросимптоматика, оживление глубоких рефлексов. Психика: характерологические особенности по типу непостоянства, поверхностности, недостаточной критичности, недооценки заболевания. Диагностика Основывается на типичных клинических проявлениях. На ЭЭГ обычно хорошо выраженный и широко распространенный альфа-ритм, иногда заостренные волны или комплексы множественных пиков или множественные пик-волны. Нет непосредственной корреляции между ЭЭГ пиками и подергиваниями. Часто наблюдается повышенная светочувствительность. Дифференциальная диагностика Эпилептические миоклонии возникают при разнообразных заболеваниях, о чем указано в разделе диффдиагностики миоклонической эпилепсии детского возраста. Важным в диагностике является учет данных ЭЭГ, возраста начала заболевания, что облегчает диффдиагностку с доброкачественной миоклонической эпилепсией детского возраста, синдромом Леннокса — Гасто, миоклонической эпилепсией Унферрихта — Лундборга, синдромом Уэста. Прогноз При адекватной терапии и отрегулируемым образом жизни прогноз благоприятный. Приступы могут персистировать в зрелом возрасте «большими припадками». Почти у всех больных после отмены лечения приступы возобновляются, поэтому даже при многолетнем отсутствии припадков нельзя прекращать прием антиконвульсантов. Социальный и витальный прогнозы благоприятные. Терапия Первый выбор — Вальпроат. Второй выбор — Этосуксимид, Клоназепам, Гексамидин. Эпилепсия с миоклоническим абсансом (синдром Тассинари) (G40.4) Этиология Данный синдром относится к криптогенным формам эпилепсии. Распространенность Встречается крайне редко, в основном у мальчиков. Клиника Возраст начала в 4—9 лет, в среднем — 7 лет. Приступы клинически характеризуются нарушением сознания по типу абсансов, которые сопровождаются тяжелыми двусторонними ритмическими клоническими (абсансы с миоклониями плечевого пояса) подергиваниями, часто сочетающимися с тоническими сокращениями. Припадки наблюдаются несколько раз в день, осознавание подергиваний может быть сохранено. Сочетанные припадки бывают редкими. Неврология: без грубых органических нарушений. Психика: в основном психомоторное развитие соответствует возрасту, но с развитием заболевания возможно отставание. Диагностика ЭЭГ: всегда двусторонние синхронные и симметричные разряды ритмических пик-волн 3 Гц, так же как и при типичных абсансах. Дифференциальная диагностика Проводится с другими формами эпилепсии, сопровождающимися абсансами. Основную роль в диффдиагностике играют данные ЭЭГ — корреляция приступов с типичной картиной абсансной активности. Учитывая, что типичные абсансы наблюдаются почти исключительно в детском возрасте, важным для диагностики является возраст их начала. Прогноз В отношении приступов и психического развития является куда менее благоприятным, чем при пикнолепсии, в связи с довольно выраженной резистентностью припадков к терапии, умственным отставанием и возможным переходом в другие виды эпилепсии, например в синдром Леннокса — Гасто. Нередко миоклонии вообще не поддаются лечению. Терапия Первый выбор: Этосуксимид, Вальпроат. Второй выбор — Карбамазепин, Бензодиазепины. Рекомендуется сочетание Вальпроата и Этосуксимида, Вальпроата и Ламотриджина, Вальпроата и Клобазама, применение новых генераций АК — Фелбамат, Габапентин, Вигабатрин. Эпилепсия с миоклонически-астатическими приступами Этиология Часто генетическая предрасположенность. У 37% больных выявлено семейное заболевание этой формой. Распространенность Встречается редко, мальчики поражаются чаще, чем девочки. Клиника Возраст начала между 7 мес. и 6-м годом жизни, обычно 2 — 5 лет. Приступы на фоне правильного психомоторного развития начинаются обычно с фебрильных или афебрильных тонико-клонических приступов, малых атонических, миоклонических, миоклонически-астатических припадков и сложных абсансов. Часто припадки идут в виде статусоподобных серий. Бессудорожные приступы составляют 36% всех случаев. В приступе бывает и тонический компонент, и даже чистые тонические приступы, но они возникают на поздних стадиях заболевания, и обычно в неблагоприятных ситуациях. Это отличает данную форму от синдрома Уэста, которому они как раз свойственны. Неврология: обычно без грубых органических нарушений. Психика: в 50% случаев психомоторное развитие соответствует возрасту. Морфология: без структурных нарушений. Диагностика Проводится с учетом этиологии, клиники приступов. ЭЭГ в начале болезни — без особенностей или с преобладанием тета-ритма. С развитием болезни — на нормальном или умеренно измененном фоне, нерегулярные комплексы пик-волна и полипик-волна 3—4 Гц. Может напоминать картину при синдроме Леннокса — Гасто, но с менее выраженной дезорганизацией и тенденцией к генерализованным, регулярным комплексам пик-волна. Выражена фотосензитивность. Фокальные и мультифокальные проявления обычно отсутствуют. Дифференциальная диагностика При неэффективности терапии, снижении познавательной способности следует провести диффдиагностику с идиопатическим синдромом Леннокса — Гасто, с миоклоническими формами детской эпилепсии. Прогноз Более благоприятный, чем при синдромах Уэста и Леннокса — Гасто, но не более чем в 50% всех случаев. Терапия Первый выбор — Вальпроат, Этосуксимид, Ламотриджин. Начинать лечение следует с Вальпроата. Второй выбор — Бензодиазепины, Клобазам, Клоназепам. При присоединении ГТКП, особенно при резистентности к терапии — бромиды, применять АКТГ. Респираторные аффективные судороги Эти пароксизмальные состояния чреваты ошибочной диагностикой. Дифференциация между респираторными аффективными приступами и эпилепсией строится на анамнестических данных и связи приступов с эмоциональными реакциями на фоне невротических проявлений. В отличие от эпилепсии, при этих приступах характерны провоцирующие факторы, крик перед судорогами, цианоз, появляющийся до судорог, опистотонус при нормальной ЭЭГ, хотя даже выявленная патология на ЭЭГ не решает вопроса дифференциации. АК при респираторных аффективных судорогах безрезультативны. Лечение должно быть направлено на устранение невротизирующих факторов. Фебрильные судороги Этиология и патогенез До сих пор нет общепринятого мнения о природе этих приступов. Существует мнение, что гиперпирексия провоцирует идиопатическую эпилепсию и Фебрильные судороги являются нередко результатом не столько экстрацеребральных, сколько церебральных процессов. Считается, что гиперпирексия является провоцирующим моментом в вызывании судорожного припадка на благоприятной для этого почве (перинатальная патология — до 50%, инфекции, травмы — до 20% и др.). Нередко в семьях обнаруживаются случаи аналогичных приступов. Распространенность Встречаются до 15% в общей популяции. Среди 55% детей, перенесших «беспричинные» детские судороги, у 27%, перенесших фебрильные судороги, наблюдается эпилепсия. Клиника Приступы тонико-клонических судорог (всегда первично-генерализованных) строго связаны с возрастом, спонтанно прекращаются в 4 — 5 лет, развиваются только при высокой температуре. Продолжительность приступа не более двух минут. Наблюдаются у детей не более 4 — 5 раз, чаще только в исключительных случаях. Затяжные фебрильные судороги могут стать причиной склероза аммонова рога с риском развития фокальной эпилепсии. Диагностика Основывается на типичности клиники, этиологии и данных ЭЭГ. Дифференциальная диагностика Дети нуждаются в тщательном исследовании и контроле. О возможности развития эпилепсии следует думать, если имеются указания в анамнезе на неврологические повреждения, фокальное начало приступов, и/или фокальности на ЭЭГ, а также при наличии более 4 — 5 приступов, при их появлении при температуре менее 38,5 град. С и при семейной предрасположенности к эпилепсии. Прогноз При первичных проявлениях в возрасте 3 лет рецидивы встречаются крайне редко. Почти у 30% детей с фебрильными судорогами в дальнейшем развивается эпилепсия. Терапия При наличии доброкачественных форм нет необходимости в применении АК терапии. При затяжных фебрильных судорогах требуется целенаправленное применение АК и другое этиопатогенетическое лечение. Синдром Леннокса — Гасто Этиология и патогенез Относится (как и синдром Уэста) к мультифакторным эпилепсиям, то есть имеются подозрения на наличие симптоматической этиологии, но не подкрепляющиеся результатами морфологических исследований, и этиология в таком случае остается криптогенной. В Международной классификации эпилепсия выделена в разделе Генерализованные формы эпилепсии как криптогенная и симптоматическая. Нередко прослеживаются органические резидуальные церебральные синдромы (пре-, пери- и постнатальные), подострые энцефалопатии, нейрометаболические заболевания, туберозный склероз. Распространенность Проявляется у детей от 2 до 8 лет, но чаще в дошкольном возрасте, 2—6 лет. Примерно 30% таких приступов рекрутируются из случаев синдрома Уэста. Клиника Начало в возрасте от 2 до 8 лет, поздние формы от 10 до 20 лет. Наиболее часты виды припадков — миоклонико-астатические припадки, атипичные абсансы, молниеносные кивательные судороги, внезапные падения, тонические приступы (обычно во сне). Нередко встречаются и генерализованные тонико-клонические, миоклонические, парциальные припадки. Имеется тенденция к серийности разнообразных приступов с состоянием ступора, а также незаметному переходу в эпилептический статус. Неврология: в 40% случаев — церебральные парезы и гипотонико-астатические нарушения. Психика: обычно — умственная отсталость до степени тяжелой деменции, психоорганические нарушения. В 80% случаев — тяжелые когнитивные и личностные нарушения органического типа. Нейрорадиология и патоморфология: фокальные или диффузные структурные нарушения. Диагностика Основывается на типичной клинической картине и ЭЭГ данных. На ЭЭГ обычно имеются изменения фона в виде медленных пик-волн меньше 3 Гц, ночью серии пиков (доходит до 100 за ночь), часто — мультифокальные изменения. Ранее считалось, что для синдрома Леннокса — Гасто патогномонична картина ритмических комплексов «пик-волна» 2,5 Гц. На самом деле описание паттерна ЭЭГ при синдроме Леннокса — Гасто это та же гипсаримия, только с большим содержанием «острых» феноменов. Заключение по ЭЭГ о гипсаритмии подтверждает диагноз синдрома Леннокса — Гасто. Дифференциальная диагностика Синдром Уэста. Прогноз В 75% случаев — резистентность к терапии. Возможно персистирование миоклонико-астатических припадков во взрослый возраст, переход в большие судорожные приступы. Неблагоприятные прогностические признаки — предшествующее органическое поражение мозга или синдром Уэста, распространенные и частые тонические судороги, наклонность к статусному течению. Лечение Обычно приступы купируются с трудом. Более чем в половине случаев синдром развивается на фоне предшествующей энцефалопатии, но в 40% случаев возникает как бы первично. Препараты первого выбора — Вальпроат, Этосуксимид. Второй выбор — Бензодиазепины, АКТГ, кортикостероиды. В последние годы препаратом первого выбора становятся Вигабатрин и Ламотриджин, которые избирательно увеличивают содержание в мозге тормозного нейротрансмиттера — ГАМК. Салаамов тик В изолированном варианте — в виде ритмических движений головой в передне-заднем направлении, к которым присоединяются кивательные движения туловища в том же направлении и, иногда, с нистагмом. Эти движения медленные и возникают у лиц с интеллектуальной недостаточностью (олигофренов), особенно в сидячем положении, сериями по 20—30. В таком варианте эти состояния не имеют отношение к эпилепсии. Салаамов тик следует отличать от салаамовых приступов, которыми обозначаются инфантильные (младенческие) спазмы или пропульсивные припадки при синдроме Уэста. При этом синдроме судороги в виде флексорных туловищных движений или даже более простых движений — «кивки», «клевки», «поклоны», «складывания» — по типу «перочинного ножа» (во франкоязычной литературе). Это как бы рудиментарные судороги, но именно судороги, насильственный поклон, а не падение головы вперед из-за утраты тонуса. Такая картина получается из-за незрелости механизмов кортико-спинального контроля. ЭЭГ в более раннем возрасте — больше разрядов дельта-активности, а в более позднем возрасте, когда нейросинаптические медиаторные системы уже обеспечивают генерацию, — больше быстрых эпилептических феноменов. Симптоматическая ранняя миоклоническая энцефалопатия (ранняя младенческая эпилептическая энцефалопатия с паттернами «вспышка-угнетение» [burst-suppression] на ЭЭГ, синдром Отахара) Этиология Заболевание относится к симптоматическим генерализованным эпилепсиям неспецифической этиологии. Часто встречаются семейные случаи заболевания, что указывает на какое-то нарушение метаболизма. Распространенность Синдром описан в 1976 году, определяется по очень раннему началу болезни, встречается редко. Клиника Начало в первые несколько месяцев жизни частым фрагментарным миоклонусом. Затем — парциальные припадки, массивные миоклонусы или тонические спазмы. Диагностика Основывается на клинических особенностях и ЭЭГ данных. На ЭЭГ — супрессивно-взрывчатая активность, которая может перейти в гипсаритмию. Дифференциальная диагностика Синдром Уэста. Прогноз Неблагоприятный. В возрасте 4—6 месяцев часто отмечается переход в синдром Уэста. Течение очень тяжелое и быстрое драматическое развитие. Психомоторное развитие прекращается и на первом же году может наступить смерть. Терапия Выраженная резистентность к лечению АК. Синдром Уэста (эпилепсия с судорогами типа молниеносных «салаам»-поклонов, «инфантильные спазмы», пропульсивные припадки) Этиология Относится (как и синдром Леннокса — Гасто) к мультифакторным эпилепсиям. В Международной классификации эпилепсии выделена как криптогенная и симптоматическая в разделе «Генерализованные формы эпилепсии». Нередко прослеживаются органические резидуальные церебральные синдромы (пре-, пери- и постнатальные), подострые энцефалопатии, нейрометаболические заболевания, в 10% случаев — туберозный склероз. Синдром Уэста можно подразделить на 2 группы: симптоматическая группа — наличие предшествующих признаков поражения мозга (предшествующая приступам умственная отсталость, неврологические, нейрорадиологические изменения или другие типы припадков, или известная этиология) и идиопатическая (меньшая) группа. Распространенность Проявляется у детей возраста 3 — 7 месяцев жизни, чаще у мальчиков. Клиника Характерна триада признаков: инфантильные спазмы + задержка психомоторного развития + гипсаритмия. Один из признаков триады может выпадать. Спазмы могут быть сгибательными, разгибательными, чаще же — смешанные. Приступы в большинстве случаев заключаются во внезапно начинающемся, напоминающим испуг, генерализованном миоклонусе с рывками вверх, поднятием головы, напоминающими реакцию Моро — молниеносные (длительность — до 1 с) клонические судороги со сгибанием головы и туловища, иногда — с падением на колени. В некоторых случаях приступы проявляются также в коротком, но серийном, кивательном движении головы («кивки»). Реже эти приступы протекают как при замедленной киносъемке, чем напоминают восточное приветствие «саалам». Всегда выявляется выраженная тенденция к серийности судорог, незаметному переходу к статусу, комбинирование с большими приступами. Психика: задержка психомоторного развития. Неврология: в 80%случаев — церебральные парезы, гипотонико-атактические нарушения, микроцефалия. Нейрорадиология: в 90% случаев находят грубые структурные нарушения. Патоморфология: микроцефалия, лиссэнцефалия, пахигирия, микрогирия, глиоматоз, глобарный склероз, сосудистые мальформации. 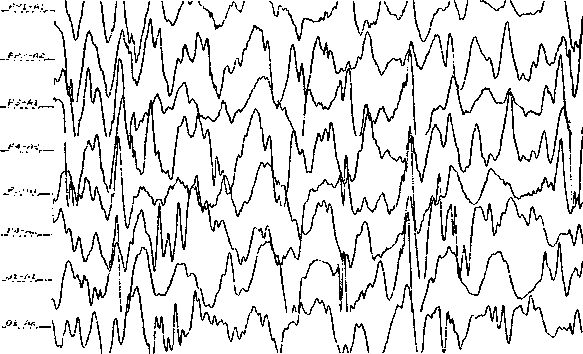 Гипсаритмия. Скорость — 30 мм/с. Амплитуда — уменьшена по сравнению с обычным усилением в 3 раза. Диагностика Основывается на типичной триаде клиники и патогномоничных ЭЭГ данных, активизирующихся во сне. ЭЭГ вне припадка — гипсаритмия. Гипсаритмия — это непрерывная генерализованная высокоамплитудная медленная и гиперсинхронная активность с острыми волнами, пиками, медленными пик-волновыми комплексами. ЭЭГ данным во время припадка при молниеносных миоклониях соответствуют генерализованные пики и острые волны, при тонических судорогах — низкоамплитудные высокочастотные генерализованные пики, нарастающие по амплитуде к концу припадка. Дифференциальная диагностика Синдром Леннокса — Гасто. Прогноз Чаще зависит от своевременного лечения АКТГ, но принципиально — от симптоматического или идиопатического характера синдрома. В основном прогноз неблагоприятный. Смертность отмечается примерно в 20% случаев. В 90% случаев — нарушение психического развития. Часто переход в синдром Леннокса — Гасто. Благоприятные прогностические признаки: нормальное психомоторное развитие к началу приступов, отсутствие других эпилептических проявлений, нормальный неврологический и нейрорадиологический статус, быстрый ответ на терапию и отсутствие рецидивов, отсутствие фокальных или мультифокальных проявлений на ЭЭГ после исчезновения гипсаритмии. Большинство идиопатических случаев показывают благоприятный прогноз, если лечение начато своевременно. Терапия На первом этапе — высокие дозировки витамина В6, что проявляется уже в первые дни лечения. При неэффективности — Вигабатрин в высоких дозах. Если нет эффекта в течение 2 недель — начать прием Вальпроата, кортикостероидов. Побочные эффекты значительны (синдром Кушинга, нефролитиаз и др.). Эпилепсия парциальная постоянная (Кожевникова) (G40.5) Этиология Кожевников описал 2 синдрома. Первый — собственно эпилепсия Кожевникова (epilepsia partialis continua) детского возраста, в этиологии которой лежит деструктивное локальное поражение мозга любой этиологии (опухоль, сосудистое, глиоз). Второй — хроническая прогредиентная Epilepsia Partialis continua (синдром Кожевникова детского возраста, синоним — синдром прогрессирующей энцефалопатии Расмуссена), где этиология вирусно-воспалительная. При собственно эпилепсии Кожевникова (epilepsiapartialis conlinua) детского возраста Клиника Представлена парциальной непрогредиентной роландической эпилепсией у детей или у взрослых, связанной с повреждением моторной коры. Возраст начала любой. Приступы: фокальные моторные припадки, длящиеся в течение дней, недель, месяцев. Психика: без особенностей. Нейрорадиология: соответствующие этиологическому фактору морфологические изменения. Неврология: клинические проявления соответствуют поражению коры и не имеют тенденции к прогрессу (резидуальная органика); прогрессирование указывает на опухоль. Диагностика Основывается на клинических проявлениях и ЭЭГ данных. На ЭЭГ — ограниченные эпилептиформные разряды в роландической области, контрлатеральной стороне судорожных проявлений. Дифференциальная диагностика Проводится на ранних этапах — с роландической эпилепсией, в дальнейшем — с локализованными симптоматическими формами. Прогноз Непрогредиентное течение, если не прогрессирует этиологический фактор. Терапия Первый выбор — Карбамазепин. Второй выбор — Вальпроат, бензодиазепины (клоназепам, клобазам). Хроническая прогредиентная Epilepsia Partialis continua (синдром прогрессирующей энцефалопатии Расмуссена) Клиника Приступы имеют начало с фокальных моторных с последующим присоединением локальных миоклоний. Вначале приступы четко фокальны, затем локально непостоянны, тенденция к генерализации. Часто припадки во сне. Возраст начала 2—10 лет. Неврология: с развитием заболевания развивается прогредиентный гемипарез. Психика: нарастание деменции, задержки психического развития. Нейрорадиология: деструктивные изменения контрлатерально гемипарезу. Диагностика Основа — клиника и ЭЭГ данные. На ЭЭГ выявляются преимущественно диффузные дельта-волны с преобладанием в контлатеральном неврологическому проявлению полушарии, мультифокальные высокоамплитудные спайки, острые волны, пик-волны в больном полушарии, с последующим вовлечением второго. Прогноз Прогредиентное течение с развитием тяжелых неврологических и интеллектуальных дефектов. Терапия Первый выбор АК — Карбамазепин, второй выбор — Вальпроат, бензодиазепины. Медикаментозному лечению не поддается. При четко одностороней локализации требуется оперативное лечение (гемисферэктомия). Первичная эпилепсия чтения (ЭЧ) Этиология В МКБ-10 не выделена. Редкая форма идиопатической фокальной эпилепсии с предположительной локализацией очага в теменно-височной области доминантного по речи полушария. Предполагается аутосомно-доминантное наследование при ЭЧ, и она имеет семейное накопление (до 40%). Раньше относилась к рефлекторным фотосензитивным, однако тот факт, что приступы провоцируются даже во время чтения по системе Брайля, опровергло этот взгляд. В настоящее время считается, что пусковым механизмом приступа является трансформация графем в фонематическую речь. Распространенность ЭЧ — один из самых редких эпилептических синдромов. Частота встречаемости ЭЧ варьирует у народов, использующих разные системы письменности: максимальна для систем с буквенным написанием и минимальна для систем с иероглифическим. Отмечено преобладание больных мужского пола в соотношении примерно 2:1. Клиника Начало ЭЧ обычно приходится на пубертатный период и позже. Редко возникает у детей раннего школьного возраста. Приступы возникают почти исключительно во время чтения, особенно вслух. Провокация связана с индивидуальными особенностями ситуации (содержание текста, характер артикуляции, освещенность). Наиболее частое проявление приступа — клонические подергивания в мышцах нижней челюсти, в жевательной мускулатуре, ощущения затруднения дыхания, «подавливания», или сенсорные нарушения, чаще в виде расплывающегося изображения. При продолжении чтения возможен переход в большой припадок. Психика и неврология — без особенностей. Диагностика ЭЭГ в межприступном периоде в 80% случаев регистрирует нормальную электроактивность. Фотосенситивность отмечена всего у 9% больных, зато провокация пароксизмальной активности во время чтения наблюдается почти в 80% случаев. Во время приступа обычно регистрируется билатерально-синхронная пик-волновая активность с амплитудным преобладанием в теменно-височных отделах доминантного полушария и/или генерализованные пик-волны. Прогноз В целом — благоприятный. Терапия Лечение АК оправдано, так как приступы имеют тенденцию со временем провоцироваться и другими факторами (разговор, игры, еда), даже могут стать спонтанными. Средства первого выбора — Вальпроат; второго выбора — Клоназепам. Имеются данные о хорошем эффекте блокатора кальциевых каналов — Флунаризина (Сибелиум) в качестве дополнительной терапии. Эпилептический статус (Status epilepticus, SE) (G41) Определяется как «стойкое эпилептическое состояние» с повторяющимися или непрерывными приступами, которые продолжаются более 30 минут или между которыми больной не может полностью достичь своего нормального психического и неврологического состояния. Этиология Этиологические факторы, определяющие развитие статуса, разнообразны. Статус может возникать как осложнение эпилепсии или быть ее манифестным проявлением. Основные причины возникновения эпилептического статуса без предшествующих эпилептических пароксизмов (de novo): — нейроинфекции, — острые нарушения мозгового кровообращения, — черепно-мозговая травма, — прогрессирующие заболевания ЦНС, — интоксикации. Распространенность Эпилептический статус встречается с частотой 18—20 случаев на 100 000 населения и является одним из наиболее распространенных неотложных неврологических состояний. В 50% случаев эпилептический статус возникает у детей раннего возраста. Среди больных эпилепсией статус также чаще отмечается у детей (10—25%), чем у взрослых (5%). Классификация Разновидности эпилептического статуса обозначаются в соответствии со встречающимися при нем формами приступов. Наиболее известны — статус судорожных припадков, статус малых припадков, статус сложных фокальных приступов, эпилепсия Кожевникова (G40.5), статус миоклонических приступов. Прогноз При SE представляет собой ситуацию, требующую неотложной помощи, так как связанная с ним смертность даже в настоящее время может доходить до 30—50%. Терапия «Статус является истинным кризисом болезни (эпилепсии) и в меньшей степени вероятным ее завершением, которого нужно избегать с помощью правильного лечения...» (L.P. Clare, T.P. Prout, 1903). Если ранее была диагностирована эпилепсия, то развитие SE всегда указывает на необходимость критического переосмысления стратегии применяемого медикаментозного лечения и прежде всего в тех случаях, когда не удается выяснить факторы, провоцирующие SE. Эпилептический статус Grand mal (судорожных припадков) (Тонико-клонический эпилептический статус) (G41.0) Этиология Причины, чаще встречающиеся у взрослых (цереброваскулярные процессы, лишение алкоголя, гипоксические состояния), в детском возрасте почти не играют никакой роли. У детей этиологически доминируют менинго-энцефалические инфекции, врожденные аномалии развития, последствия церебральных повреждений, прогрессирующие нейродегенеративные заболевания. У новорожденных в подавляющем большинстве случаев статусы обусловлены нейрометаболическими нарушениями, инфекциями, кровоизлияниями в мозг, гипоксически-ишемическими энцефалопатиями, а в раннем грудном возрасте — острыми воспалениями и электролитными нарушениями. У пациентов с выявленной ранее эпилепсией наиболее частой причиной статусов является снижение концентрации АК в крови (неправильная смена терапии, отмена антиконвульсантов). Наиболее частые «поставщики» статусов — лобнодолевые эпилепсии. Если статус встречается впервые, причиной может быть целый ряд основных заболеваний (опухоль мозга, энцефалит, церебро-васкулярная патология, черепно-мозговые травмы, интоксикации, алкоголизм, метаболические нарушения). Эти заболевания должны быть диагностированы и лечиться параллельно со статусом приступов. Клиника Частота судорожных приступов составляет от 3 до 20 в час. Основные критерии SE — наличие выраженных изменений, вызванных предшествующим припадком и относящихся к состоянию сознания, дыхания, гемодинамики. Сознание ко времени возникновения следующего припадка полностью не восстанавливается, и больной остается в состоянии оглушения, сопора или комы. При SE пролонгированном в клинике наступают изменения: длительность ГТКП уменьшается, коматозное состояние углубляется, судороги принимают тонический характер, гипотония мышц сменяется атонией, а гиперрефлексия — арефлексией. Нарастают гемодинамические и дыхательные нарушения. Наконец, судороги могут полностью прекратиться и наступает стадия эпилептической прострации: глазные щели и рот полуоткрыты, взор безучастный, зрачки широкие. В таком состоянии может наступить смерть. Диагностика SE изучен достаточно хорошо и его диагностика не вызывает затруднений при клиническом наблюдении. Прогноз Прогноз значительно зависит от его этиологии, так как смертность от статусов Grand mal в случаях ранее диагностированной эпилепсии — не 30—50%, как при острых симптоматических, а только около 5%. Вторым по важности прогностическим фактором является продолжительность статуса. Если статус длится более 30 минут, следует опасаться развития серьезных церебральных, сердечно-сосудистых, респираторных, вегетативных и метаболических осложнений (отек мозга, гипоксия, гипотензия, гиперпирексия, лактатацидоз, изменения электролитного баланса), которые приводят к необратимым неврологическим и нейропсихологическим нарушениям. Терапия В международной практике принято использовать унифицированную этапную схему со строго определенными временными рамками. На первом этапе применяется комбинированное лечение Диазепамом и Фенитоином, которое купирует статус больших приступов в 85— 90% случаев. Этап 1 (0—10 мин.) A) Необходимо обеспечить функции дыхания и кровообращения, при необходимости — кислородный зонд. Б) Определить концентрацию противоэпилептического препарата в крови. B) Измерить температуру. Этап 2 (30—40 мин) А) Диазепам 20 мг (детям 0,2—0,4 мг/кг/м.т.) ректально, или медленно внутривенно, или Клоназепам 2 мг (детям 0,01 — 0,04 мг/кг/ м.т.) медленно внутривенно. Следует учитывать быстрое наступление действия (5—15 мин), однако не только в плане противосудорожных эффектов, но и угнетения дыхания, седативного эффекта. Б) Сразу после А) назначается внутривенно Фенитоин (детям — 10—15—20 мг/кг/м.т.), скорость инъекции — менее 50 мг/мин. Следует учитывать, что максимальный эффект наступит через 20—30 мин. При падении АД, возникновении аритмии скорость введения необходимо уменьшать. Часто первым симптомом интоксикации является нистагм. В настоящее время на нашем рынке появляется инфузионная форма Фенитоина (например, Фенгидан фирмы «Деситин»). Имеются «альтернативные» варианты международных стандартизированных схем, причем оговаривается, что «для лечения невосприимчивого к терапии статуса не существует надежных рекомендаций». «Невосприимчивым», или резистентным (рефрактерным), считают статус, который продолжается 60 и более минут, несмотря на применение не менее двух антиконвульсантов первой очереди выбора. Эти варианты предполагают применение инфузионных форм Фенобарбитала или Лидокаина, или бензодиазепинов (Лоразепама, Паральдегида). Фенобарбитал (детям: 4—6—10 мг/кг/м.т.) вводится внутривенно, скорость введения — менее 100 мг/мин. Следует учитывать возможность угнетения дыхания, седативный эффект, большой период полувыведения из организма. Лидокаин — инъекция вводятся ударной дозы 100—200 мг внутривенно, затем — инфузия 3—4 мг/кг. Следует учитывать возможность аритмии, падения АД, реакции идиосинкразии, немедленное действие. Лоразепам 4 мг (+4 мг через 10 минут) вводится внутривенно. Следует учитывать возможность угнетения дыхания, седативный эффект, длительность действия около 12 часов. Этап 3 (рефрактерный статус) Применяется общая анестезия (наркоз), например с помощью тиопентала, который проводится в отделении интенсивной терапии (реанимации). Наркоз необходимо продолжать 12—24 часа после последнего приступа. Лучше, конечно, постоянно регистрировать ЭЭГ с целью подавления «вспышек». Эпилептический статус Petit mal (Эпилептический статус абсансов, SEA) (G41.1) Этиология Эта форма статуса может быть первым проявлением эпилепсии, при котором у пожилых людей внезапно развивается спутанность сознания. Статус абсансов может последовать за «большим» приступом или перейти в него. Распространенность Наиболее часто встречается у детей. Может возникать в 5% случаев генерализованных эпилепсии. Клиника SEA представляет собой разновидность генерализованного бессудорожного статуса. Состояние известно под устаревшими терминами: «статус спутанности», «пик-волновой ступор». Имеющиеся нарушения сознания выражены в различной степени — от легкого нарушения концентрации до дезориентации и ступора. В отдельных случаях изменение сознания столь незначительно, что может быть выявлено только при психологическом тестировании. Примерно у 50% больных наблюдается дрожание век, подергивание рук и другие судорожные проявления. SEA в ряде случаев является причиной эпилептической фуги. Различают две разновидности — статус типичных и статус атипичных абсансов — по ряду признаков, главным из которых являются ЭЭГ проявления. Для статуса «типичных» абсансов клинически характерны: — внезапное начало и окончание, — продолжительность — до нескольких дней (обычно — меньше), — трансоподобное состояние с отсутствием реакций на внешние стимулы, — наличие в анамнезе идиопатических форм генерализованных эпилепсии. Для статуса «атипичных» абсансов клинически характерны: — постепенное начало и окончание, наличие продромального периода, — продолжительность — до нескольких недель, — сочетание атипичных абсансов с тоническими, миоклоническими приступами, — наличие в анамнезе симптоматических или криптогенных генерализованных эпилепсии (чаще — синдрома Леннокса — Гасто). Диагностика Основывается на клинике и обязательном ЭЭГ исследовании. ЭЭГ всегда показывает комплексы пик-волн, более или менее непрерывные. Частота разрядов и морфология комплексов может часто отличаться от их классического рисунка — 3 в с. Для статуса «типичных» абсансов характерны на ЭЭГ — наличие генерализованных билатерально-синхронных комплексов «пик-волна», регулярно повторяющихся с частотой 3 в с. Для статуса «атипичных» абсансов на ЭЭГ — наличие продолжительных медленных комплексов «пик-волн», нерегулярно повторяющихся с частотой 1,5—2 Гц. Терапия Вводятся в/в инъекции препаратов группы бензодиазепинов — Диазепам в дозе 10—20 мг, детям — 0,02 — 0,04 мг/кг/м.т., или ректальное введение 20—30 мг Диазепама. Грудным детям — 5 мг, детям с массой тела более 15 кг — 10—20 мг. В последнее время применяют также в/в введение Вальпроатов. Терапия статуса абсансов должна проводиться по возможности при продолжающемся ЭЭГ контроле. |