Инсерции и делеции в уникальных и повторяющихся последовательнос. Уникальные днкпоследовательности генома человека
 Скачать 394.25 Kb. Скачать 394.25 Kb.
|

|
В ходе реализации проекта «Геном человека» стало ясно, что организация ДНК в человеческом геноме значительно более изменчива, чем считалось раньше. Из 3 млрд пар оснований ДНК в геноме только около половины состоит из так называемой однокопийной, или уникальной, ДНК, т.е. ДНК, нуклеотидная последовательность которой представлена однократно (или только несколько раз). Остальная часть генома состоит из нескольких классов повторяющейся ДНК и включает ДНК, нуклеотидная последовательность которой повторяется или полностью, или с некоторыми вариациями, от сотен до миллионов раз в геноме. Поскольку большинство (хотя и не все) из предполагаемых 30 000 генов в геноме представлены единственной копией, повторяющиеся части ДНК способствуют поддержанию хромосомной структуры и в значительной мере обеспечивают вариабельность между различными индивидуумами. Некоторые из таких вариантов могут предрасполагать к патологическим событиям в геноме. Уникальные ДНК-последовательности генома человека Хотя уникальные ДНК-последовательности занимают по крайней мере половину генома, большинство их функций остается загадкой, поскольку последовательности, действительно кодирующие белки (т.е. кодирующие части генов), составляют только небольшую часть всей уникальной ДНК. Большинство уникальной ДНК обнаруживают в виде коротких участков (несколько килобаз и даже короче), перемежающихся с участками повторяющейся ДНК различных типов. Повторяющиеся последовательности широко представлены в любом организме. В таблице видно, как часто повторяющиеся последовательности могут встречаться у некоторых организмов. 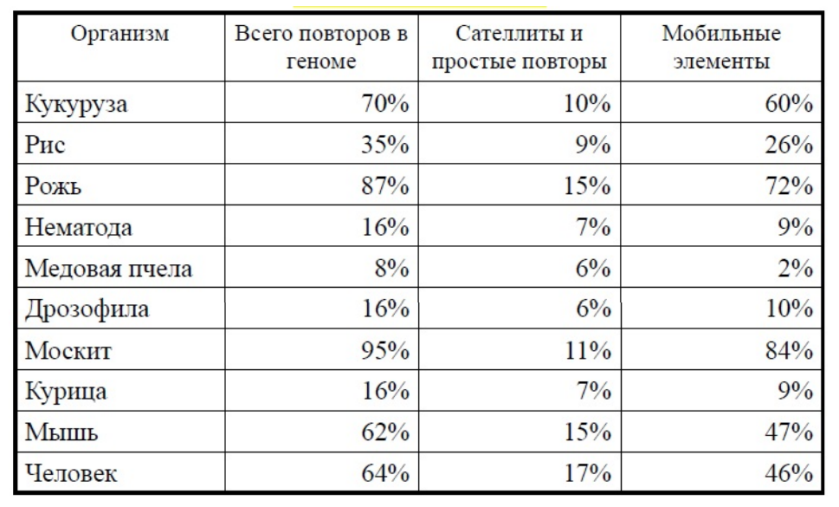 Повторяющиеся последовательности в ДНК некоторых организмов К повторяющимся элементам относят тандемные повторы и диспергированные повторы. Тандемные повторы тоже можно разделить на: • сателлиты — длина повторяющегося слова более 100 нуклеотидов. • минисателлиты — повторы из слов длиной от 7 до 100 нуклеотидов. • микросателлиты — самые маленькие повторы по 1 – 6 нуклеотидов. Тандемные повторы возникают за счет ДНК-полимеразы, которая при репликации этих участков зависает и синтезирует еще несколько повторов. Или синтезирует меньшее количество повторов. Это происходит довольно часто. В каких-то случаях это может приводить к генетическим заболеваниям. При умножении полиглутаминовых трактов (GAA, GAG) может развиться болезнь Гентигтона, болезнь Кеннеди, спиноцеребеллярная атаксия типа 1, 2, 3, 6, 7, 17. При умножении неполиглутаминовых трактов — синдром ломкой X-хромосомы, атаксия Фридрейха, спиноцеребеллярная атаксия типа 8, 12. Механизм возникновения этих заболеваний теперь известен. Повторы могут возникнуть в промоторе (приводит к изменению регуляции гена), экзоне ( — изменение белка), интроне ( — изменение транскрипции). Синдро́м Ма́ртина — Белл (синдром ломкой X-хромосомы, fragile X syndrome, FraX (от англ. fragile — хрупкий, ломкий)) — наследственное заболевание. Развитие синдрома связано с экспансией единичных тринуклеотидов (ЦГГ) в Х-хромосоме и приводит к недостаточной экспрессии белка FMR1, который необходим для нормального развития нервной системы. Существует четыре основных состояния хромосомного участка, подверженного нарушениям при синдроме ломкой Х-хромосомы, которые относятся к удлинению повторяющихся последовательностей ЦГГ. Нормальное количество повторов (отсутствие синдрома) — от 29 до 31. Премутация — от 55 до 200 повторов (синдром не развивается). Полная мутация — более 200 повторов (обычно от 230 до 4000), при которой проявляется синдром. Промежуточное состояние, или аллели серой зоны, — от 40 до 60 повторов[2]. Синдром ломкой Х-хромосомы развивается в результате мутации гена FMR1 в Х-хромосоме. Мутация в этом гене встречается приблизительно у одного из 2000 мужчин и у одной из 259 женщин. Распространённость непосредственно заболевания — приблизительно 1 из 4000 мужчин и 6000 женщин[4]. Экспансия повторяющихся кодонов ЦГГ приводит к гиперметилированию ДНК в промоторе гена FMR1 и, как следствие, фактическому прекращению его экспрессии. Как предполагают, аномальное метилирование промотора гена FMR1 в локусе Хq27.3 является причиной формирования сайта ломкости Х-хромосомы. По этому цитогенетическому признаку синдром Мартина — Белл получил своё второе название — синдром ломкой Х-хромосомы. Мутация гена FMR1 приводит к подавлению транскрипции белка FMR1. У здоровых индивидов FMR1, как считают, регулирует значительную популяцию мРНК: FMR1 играет важную роль в обучении и запоминании, а также принимает участие в развитии аксонов, формировании синапсов, появлении и развитии нервных связей[5]. Болезнь Гентингтона (синдром Гентингтона, хорея Гентингтона) — аутосомно-доминантное генетическое заболевание нервной системы, характеризующееся постепенным началом обычно в возрасте 30—50 лет и сочетанием прогрессирующего хореического гиперкинеза и психических расстройств. Заболевание вызывается умножением кодона CAG в гене HTT. Этот ген кодирует 350-kDa белок гентингтин с неизвестной функцией. В гене дикого типа (не мутантного) у разных людей присутствует разное количество CAG-повторов, однако, когда число повторов превышает 36, развивается болезнь. Нейроморфологическая картина характеризуется атрофией стриатумa, а на поздней стадии также атрофией коры головного мозга. Ген HTT, присутствующий у всех людей, кодирует белок гентингтин (Htt). Ген HTT расположен на коротком плече 4-й хромосомы (4p16.3)[6]. Этот ген содержит в себе участок с повторяющейся последовательностью трёх азотистых оснований — цитозин-аденин-гуанин (то есть, ЦАГЦАГЦАГ…). Триплет ЦАГ кодирует аминокислоту глутамин, поэтому синтезируемый белок гентингтин содержит последовательность глутаминовых аминокислот, называемую полиглутаминовый тракт[7]. Количество ЦАГ триплетов различно у отдельных лиц и может изменяться с последующими поколениями. Если их становится больше 36, то синтезируется удлинённый полиглутаминовый тракт и происходит образование мутантного белка гентингтина (mHtt)[8], который оказывает токсичное действие на клетки и вызывает болезнь Гентингтона. Как правило, от числа ЦАГ повторов зависит степень повреждений, наличие около 60 % повторов сверх нормы вызывает появление симптомов в различном возрасте[6]. 36—40 повторов приводят к редуцированной пенетрантности формы этого заболевания, которая намного позже проявляется и медленнее прогрессирует. В некоторых случаях начало болезни может быть настолько поздним, что симптомы никогда не обнаруживаются[9]. При очень большом количестве повторов болезнь Гентингтона имеет полную пенетрантность и может проявиться до 20 лет, тогда болезнь классифицируется как ювенильная, акинетически-ригидная или Вестфаль варианты. Составляет приблизительно 7 % случаев болезни Гентингтона[10]. Мутантный ген был предположительно завезён в США в 1630 году двумя братьями, эмигрировавшими из Эссекса в Бостон[11][12]. Болезнь передаётся по наследству. Мутантный аллель доминантный, поэтому в семье, где один из родителей несёт такую мутацию, каждый из потомков может получить её с вероятностью 50 %. Наследование не зависит от пола носителя или его детей. . Минисателлитные и микросателлитные повторы могут совместно обозначаться как локусы с варьирующим числом тандемных повторов (variable number of tandem repeat locus, VNTR). Изучать их нетрудно: детектирование VNTR возможно при помощи доступных молекулярных методов. Благодаря этому можно выявить число тандемных повторов в определенном гене любого из нас — причем их может оказаться и 3, и 15 без заметных отличий в функционировании этого гена. Известна значительная изменчивость кратности повторов, то есть количества копий повторяющейся единицы последовательности: это связано с тем, что тандемные повторы являются «горячими точками» мутагенеза. Вероятность возникновения «ошибок» в таких текстах превосходит вероятность стандартных «опечаток» (точечных мутаций «неповторимой» ДНК) примерно в 100 000 раз. Причина этого связана с так называемыми ошибками репликации при редактировании (slipped strand mispairing). Поскольку тандемные повторы представляют собой расположенные одна за другой идентичные последовательности, две комплементарные цепочки дуплекса ДНК могут принять неправильное расположение друг относительно друга. При этом происходит как бы проскальзывание одной цепи относительно другой (от англ. to slip — «проскользнуть», «сдвинуться») (рис. 1). 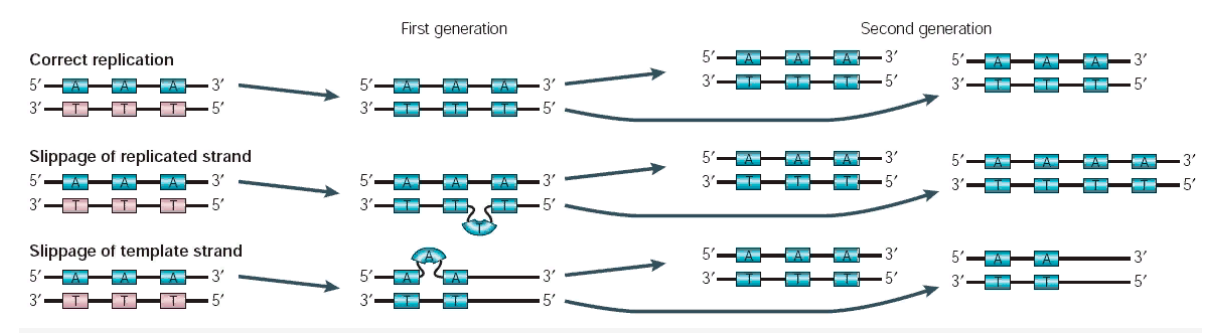 Ошибки репликации при редактировании (slipped strand mispairing) способны вызвать изменение экспрессии гена. Неправильная гибридизация цепей ДНК в области тандемных повторов при репликации и ошибки системы репарации (нужной для исправления таких случаев) могут привести к вставке (экспансии) или удалению (делеции) одного или более повторов. В ходе репликации первый повтор одной цепи, скажем, GTAC, может оказаться спаренным с первым повтором CATG другой цепи, но может также быть связан и со вторым, третьим и т.д. Некоторые последовательности-повторы могут быть вытеснены в сторону и в результате этого оказаться вне транскрипции. В связи с этим нарушением комплементарности ферменты репликации могут допускать ошибки при синтезе копии ДНК — а именно «потерять» или добавить дополнительные копии повторов (рис. 1). В этом случае возникают специфические мутации — триплеты остаются правильными, а вот их число изменяется. Поскольку затронутой оказывается значительная часть последовательности (не единичный нуклеотид), ДНК приобретает довольно существенные изменения. Ошибкам репликации при редактировании могут противодействовать точечные мутации внутри последовательности отдельного повтора. Это справедливо даже в том случае, если произошла замена на синонимичный (то есть кодирующий ту же аминокислоту) кодон. Эти однобуквенные замены приводят к возникновению изменения, заметного на однородном фоне точных повторов. Благодаря такой метке можно будет избежать проскальзывания и в результате обе цепи дуплекса будут сориентированы друга относительно друга надлежащим образом. С течением времени и новых репликаций в таком стабильном тандемном повторе накапливаются небольшие изменения, в связи с чем он всё больше изменяется. Таким образом, последовательность тандемных повторов может постепенно исчезать. Возможен и противоположный случай, когда удаление повтора (его делеция) приводит к исчезновению имеющегося однобуквенного отличия-мутации, а последующая неправильная гибридизация — привести к синтезу точного повтора. Это предоставляет возможность использовать уровень «безошибочности» тандемного повтора для того, чтобы установить, насколько интенсивному отбору он подвергался в прошлом. Если неточностей окажется немного, это свидетельствует о многочисленных вставках (называемых в этом случае экспансиями) и выпадениях (делециях). Диспергированные повторы, или мобильные элементы — это последовательности, которые могут перемещаться по геному. Их тоже делят на два класса: • транспозоны с помощью ферментов транспозаз вырезаются и целиком вставляются в другое место генома. Они составляют 2 – 3% генома человека. • ретротранспозоны Их перемещения осуществляется с участием обратной транскрипции. Ретротранспозон транскрибируется, с его РНК синтезируется кДНК. Эта копия может встроиться в любое место генома. Ретротранспозоны составляют 42% генома. Самый известный ретротранспозон носит название Alu повтора. Инсерции Alu-повторов в экзоны и промоторы белок-кодирующих генов, в область экзон-интронных границ, а также незаконная гомологичная рекомбинация между различными копиями Alu-повторов могут приводить к возникновению различных наследственных заболеваний. Многие факты свидетельствуют о том, что Alu-повторы могут участвовать в регуляции генной экспрессии, как на уровне транскрипции, так и на уровне трансляции. Так, например, Alu-повторы встречаются в 3'-нетранслируемой области некоторых мРНК и могут существенно влиять на их стабильность. Alu-повторы содержат в своем составе множественные CpG-динуклеотиды, являющиеся сайтами метилирования в геноме. Как известно, степень метилирования цитозина в последовательностях 5'-CG, фланкирующих 5'-область гена, коррелирует с уровнем экспрессии генов. Однако в большинстве случаев инсерции Alu-повтора являются только маркерами болезней, и присутствие определённого аллеля не означает, что её носитель обязательно будет иметь данную болезнь. Впервые о связи опосредованной Alu-повтором рекомбинации с наследственной предрасположенностью к раку было сообщено в 1995 году. С инсерциями Alu-повторов могут быть связаны следующие заболевания человека[7]: рак молочной железы саркома Юинга семейная гиперхолестеринемия гемофилия нейрофиброматоз сахарный диабет II типа Уникальные последовательности в геноме – те последовательности, в частности – белоккодирующие, которые представлены в геноме один или два раза. Для них тоже характерны инсерции и делеции. В данном случае они будут приводить к сдвигу рамки считывания, если число вставившихся или элиминирующих нуклеотидов будет не кратно трем. Делеции: Делеции и инсерции в уникальных последовательностях генома Тая–Сакса является результатом мутаций в гене HEXA на хромосоме 15, который кодирует альфа-субъединицу бета-N-ацетилгексозаминидазы А, лизосомального фермента. К 2000 году в гене HEXA человека было выявлено более 100 различных мутаций.[14] Эти мутации включали одиночные базовые вставки и делеции, мутации фазы сплайсинга, миссенс-мутации и другие более сложные паттерны. Каждая из этих мутаций изменяет белковый продукт гена (т. Е. фермент), иногда сильно подавляя его функцию.[15] В последние годы популяционные исследования и анализ родословной показали, как такие мутации возникают и распространяются в небольших популяциях основателей.: Евреи ашкенази. Вставка четырех пар оснований в экзон 11 (1278insTATC) приводит к изменению рамки считывания гена HEXA. Эта мутация является наиболее распространенной мутацией в еврейском населении ашкенази и приводит к инфантильной форме болезни Тая–Сакса[16]. Каджуны. Та же мутация 1278insTATC, обнаруженная среди ашкеназских евреев, встречается у каджунской популяции южной Луизианы. Исследователи проследили родословную носителей из семей Луизианы до одной пары основателей, которые, как известно, не были евреями, которые жили во Франции в 18 веке. Французские канадцы. Две мутации, не связанные с мутацией ашкенази/каджуна, отсутствуют во Франции, но распространены среди некоторых франко-канадских общин, проживающих в юго-восточном Квебеке, и акадийцев из провинции Нью-Брансуик. Анализ родословной показывает, что мутации были редки до конца 17 века. Муковисцидоз: В 1989 году был найден ген CFTR , кодирующий хлорный канал длиной 1480 аминокислот, и начат поиск мутаций, отвечающих за развитие муковисцидоза. Всего описано более 2000 мутаций в гене CFTR, однако только 250–300 из них приводят к муковисцидозу, а достаточно часто встречается (более чем у 0,1% больных) примерно 20 [11]. Мутации I классаМутации I класса встречаются примерно у 10% пациентов. При них белок CFTR вообще не синтезируется или синтезируется в усеченном виде и сразу деградирует, потому что в гене произошла замена кодирующего аминокислоту кодона на стоп-кодон, или сдвиг рамки считывания, или появился сигнал неправильного сплайсинга. Самая частая мутация — замена глицина-542 на стоп-кодон. Мутации II классаНаиболее распространены мутации II класса, вызывающие неправильные сворачивание белка и последующий процессинг клеточными механизмами. Самая частая мутация — F508del (делеция фенилаланина в положении 508 ). 70% пациентов гомозиготны по этой мутации (то есть она присутствует в обеих копиях гена CFTR), а у 90% есть хотя бы один мутантный аллель [14]. У гомозиготных пациентов наблюдается тяжелое течение муковисцидоза, а гетерозиготы по CFTR-F508del, у которых одна из копий гена нормальна, не имеют симптомов болезни. Существует гипотеза, объясняющая стабильность такого тяжелого заболевания в человеческой популяции: у гетерозигот в меньшей степени происходит потеря воды при болезнях, сопровождающихся диареей, например, при холере и брюшном тифе. Соответственно, раньше, когда эти болезни были одной из основных причин смертности, особенно детской, шел отбор на дефектные копии гена [15]. F — обозначение фенилаланина, а del обозначает делецию, то есть отсутствие аминокислоты. Мутация F508del приводит к тому, что белок неправильно сворачивается и еще в эндоплазматическом ретикулуме не проходит «контроль качества» со стороны клеточных систем и направляется на деградацию, не доходя до плазматической мембраны. https://lk.msu.ru/uploads/attachments/attachment_1951_1605614321.pdf https://www.herzen.spb.ru/uploads/zhukovaaa/files/Лекция2%20и%203_Геном%20эукариот.%20Хроматин_Типы%20последовательностей.pdf https://biomolecula.ru/articles/alu-istoriia-odnoi-posledovatelnosti |