Тема 21. Фенолы и хиноны. 1. Классификация и номенклатура
 Скачать 294.18 Kb. Скачать 294.18 Kb.
|

1 2 Наиболее общий метод получения гидроксикетонов ароматического ряда основан на перегруппировке Фриса. Ариловые эфиры карбоновых кислот при нагревании с AlCl3 или AlBr3перегруппировываются в изомерные орто- и пара-гидроксикетоны. 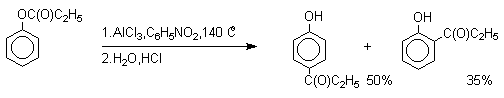 Соотношение орто- и пара-изомеров зависит главным образом от температуры и растворителя. В более жестких условиях преобладает орто-гидроксикетон, а при 20-25оС - пара-гидроксикетон. 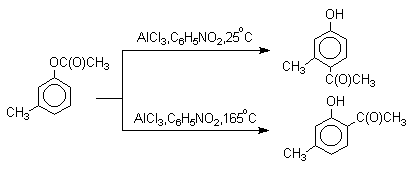 Механизм перегруппировки Фриса, по-видимому, заключается в межмолекулярном ацилировании орто- или пара-положения бензольного кольца арилового эфира комплексом второй молекулы сложного эфира и AlCl3 с образованием ацильного производного гидроксикетона и фенола. 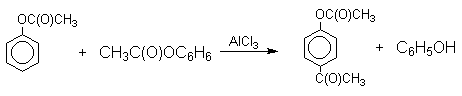 Перегруппировка завершается межмолекулярным переносом ацильной группы к фенолу.  Формилирование Формилирование – это введение группы СНО (см. лек.35). Синтетически наиболее важными являются формилирование фенолов по Вильсмейеру-Хааку и Реймеру-Тиману. Реакция Вильсмейера-Хаака N-Алкиламиды муравьиной кислоты - N,N-диметилформамид (ДМФА) и N-метилформамид в присутствии хлорокиси фосфора являются региоселективными формилирующими агентами. С помощью этих реагентов альдегидная группа вводится в пара-положение по отношению к ОН -группе. Эту реакцию можно также рассматривать как ацилирование, где роль катализатора (кислоты Льюиса) выполняют хлорокись фосфора (POCl3). Наиболее эффективна система ДМФА-POCl3, в которой ДМФА служит и реагентом, и растворителем. Электрофильным агентом в реакции Вильсмейера-Хаака является иминиевая соль, которая образуется при взаимодействии ДМФА и хлорокиси фосфора. 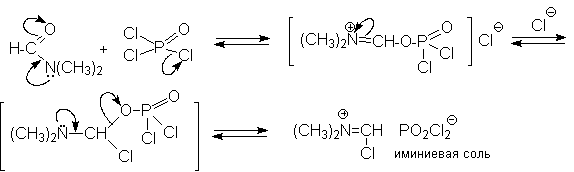 Иминиевая соль при необходимости может быть выделена в индивидуальном виде, однако обычно ее не выделяют и используют непосредственно после ее образования. 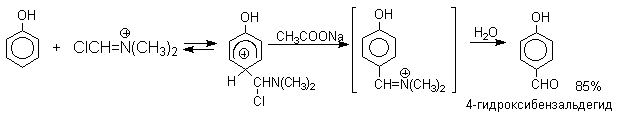 Реакция Вильсмейера-Хаака чрезвычайно проста в экспериментальном отношении и обеспечивает очень высокие выходы ароматических гидроксиальдегидов. Реакция Реймера-Тимана Формилирование фенолов по Реймеру-Тиману достигается при нагревании смеси фенола и большого избытка хлороформа с водным раствором гидроксида натрия при 50-70оС. Выходы альдегидов обычно невелики и редко превышают 30%, однако метод исключительно прост и доступен в практическом отношении. Главное достоинство реакции Реймера-Тимана заключается в преимущественном образовании орто-, а не пара-изомеров, как это имеет место в реакции Вильсмейера-Хаака. 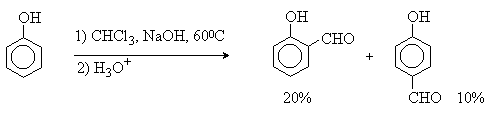 Механизм включает образование дихлоркарбена как интермедиата. Дихлоркарбен :CCl2 выполняет роль электрофильного агента по отношению к феноксид-иону, образующемуся в щелочной среде. Предполагаемый механизм реакции Реймера-Тимана может быть представлен следующей последовательностью превращений: 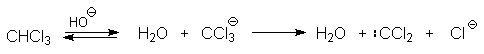 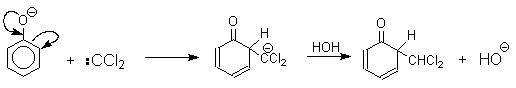 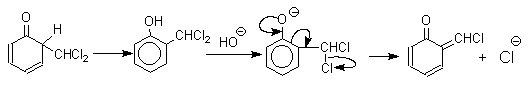 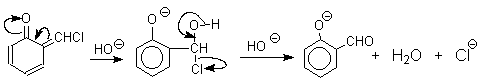 Реакция протекает только в сильно щелочной среде при наличии фенольного гидроксила, тогда как простые эфиры фенолов и диалкиланилины не формилируются в этих условиях. Карбоксилирование феноксид-ионов (реакция Кольбе) Будучи сильными нуклеофилами феноксид-анионы способны взаимодействовать с таким слабым электрофильным реагентом как оксид углерода (IV). При нагревании сухих фенолятов натрия или лития с СО2 при повышенном давлении, образуются натриевые или литиевые соли салициловой кислоты. 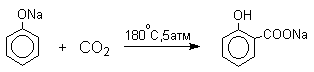 В аналогичных условиях из фенолятов калия, рубидия и цезия получаются только соли пара-гидроксибензойной кислоты. 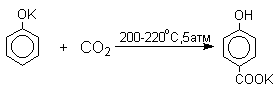 Такое различие в направлении карбоксилирования Na- и К-солей фенола принято объяснять различием в хелатообразовании этих двух катионов с атомом кислорода CO2 в переходном состоянии реакции, приводящем к салициловой кислоте. Катионы натрия и, особенно, лития значительно более эффективны по сравнению с катионом калия в способности к образованию координационной связи с атомом кислорода. 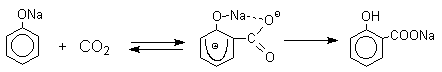 Предполагается, что для фенолятов калия, рубидия и цезия электрофильная атака осуществляется исключительно в пара-положение без какой-либо координации катиона по атому кислорода. В отличие от одноатомных фенолов двухатомные и трехатомные фенолы карбоксилируются в более мягких условиях. Так, резорцин карбоксилируется при пропускании СО2 в водный раствор его дикалиевой соли при 50оС. При этом образуется 2,4-дигидроксибензойная кислота. 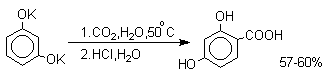 Конденсация с альдегидами и кетонами Фенолы реагируют с формальдегидом в водном растворе в присутствии основания с образованием полимерного продукта, получившего название феноло-формальдегидной смолы, карболита или бакелита. 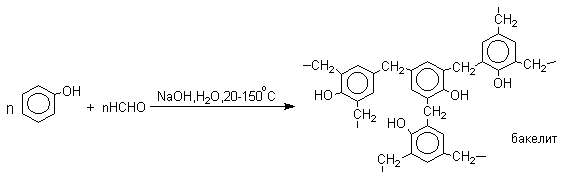 Взаимодействие феноксид-иона с формальдегидом напоминает альдольную конденсацию с той лишь разницей, что роль нуклеофильного агента вместо енолят-иона выполняет амбидентный феноксид-ион, а карбонильной компонентой является формальдегид. 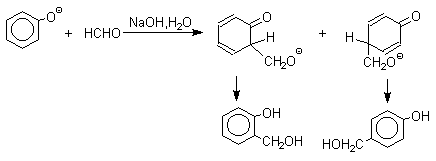 Подобно альдолям, орто- и пара-изомеры гидроксиметилфенола подвергаются дегидратации с образованием хинонметидов - соединений, родственных орто- и пара-хинонам. 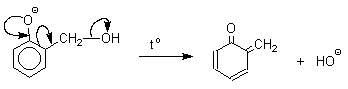 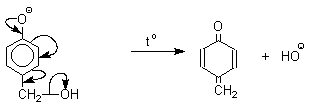 Последующее присоединение феноксид-иона к хинонметиду представляет собой присоединение амбидентного аниона к a ,b -непредельному кетону по Михаэлю. 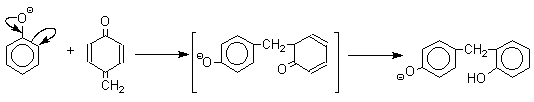 В результате дальнейшей поликонденсации в орто- и пара-положение к гидроксигруппе фенола получается трехмерная структура конечного продукта - бакелита. Бакелит представляет собой прозрачную смолу, в которой линейные звенья связаны "поперечными" связями в пара-положениях. Фенол конденсируется с ацетоном в кислой среде с образованием так называемого бисфенола А. 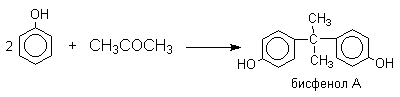 Получено много подобных продуктов конденсации фенолов с кетонами. Они находят применение в качестве антиоксидантов и мономеров для получения эпоксидных смол. Окисление Окисление фенолов относится к числу сложных, многостадийных процессов, приводящих к продуктам разного строения. Механизм окисления может сильно меняться в зависимости от строения фенола и природы одно- или двухэлектронного окислителя. Механизм окисления включает стадию образования стабилизированных резонансом ароксильных радикалов. 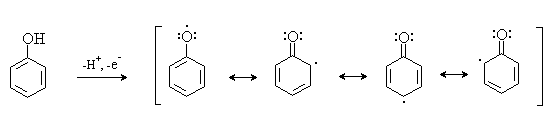 Направление дальнейших превращений зависит от характера заместителей в ароматическом кольце. При окислении большинства фенолов образуется несколько различных форм димеров в результате образования новых связей С-С между орто-орто, орто-пара- и пара-пара-положениями ароксильных радикалов, а также новых С-О связей между атомом кислорода одного радикала и орто- или пара-положением другой радикальной частицы. Всего, таким образом, образуется потенциально не менее пяти различных типов димеров, которые находится в равновесии с исходным ароксильным радикалом. Например, для монозамещенного фенола: 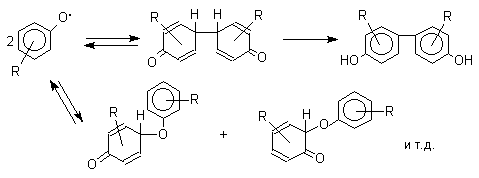 Димеры, возникающие в результате рекомбинации радикалов с образованием новой С-С связи, называются хинолидами. Хинолиды далее изомеризуются с образованием изомерных дигидроксибифенилов. Другой тип димеров, содержащих центральную связь С-О, носит название хиноловыхэфиров. Ароксильные радикалы пространственно затрудненных фенолов, содержащие в обоих орто- и пара-положении третичные алкильные группы, мономерны и не проявляют тенденции к образованию димеров в растворе. Например, при окислении 2,4,6-три-трет-бутилфенола гексацианоферратом(III) калия K3Fe(CN)6 в бинарной системе бензол-вода в инертной атмосфере образуется устойчивый радикал одновалентного кислорода - три-трет-бутилфеноксил, окрашенный в синий цвет. 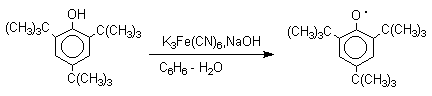 Этот радикал находится в мономерной форме в 0,1 молярном растворе в бензоле или эфире, а также в кристаллическом состоянии. Он очень чувствителен к действию кислорода воздуха, оксида азота (IY), оксида азота (II) и других радикальных частиц. Стабильность ароксильных радикалов пространственно затрудненых фенолов обусловливает их антиокислительные свойства. Они выполняют роль ловушек свободных радикалов в процессах пероксидного окисления. Активный свободный радикал, ведущий цепь окисления, быстро взаимодействует с таким фенолом, давая устойчивый ароксильный радикал, что приводит к обрыву цепи. ArOH + RO2s® ArOs + ROOH Пространственно затрудненные фенолы (ионол, гальваноксил) используют как антиоксиданты, стабилизирующие синтетические каучуки, пищевые жиры, витамины и др. ХИНОНЫ План
1. Классификация и номенклатура Хиноны по своей структуре являются циклогексадиенонами, но их названия происходят от ароматических углеводородов: бензохинон от бензола, толухинон от толуола, нафтохинон от нафталина и т.д. Цифры в начале названия обозначают положение двух карбонильных групп. 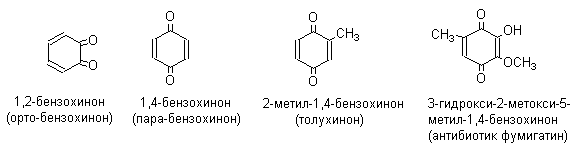 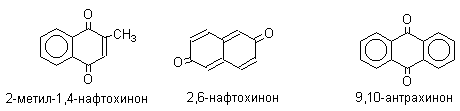  Многие производные хинонов составляют важную группу природных веществ - красителей, пигментов, антибиотиков, витаминов и т.д.  2. Методы получения Хиноны получают окислением одно- и двухатомных фенолов, аминов и диаминов ароматического ряда. Самый удобный способ получения хинонов заключается в окислении одноатомных фенолов солью Фреми - нитрозодисульфонатом калия. Эта реакция осуществляется в исключительно мягких условиях в водном спирте или ацетоне, выход обычно превышает 90%. 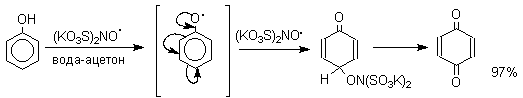 Приведенный на этой схеме циклогексадиеноновый интермедиат был выделен, что доказывает механизм одноэлектронного окисления фенолов солью Фреми. Другим одноэлектронным окислителем фенолов является карбонат серебра. Этот реагент, согласно данным последних лет, особенно пригоден для окисления 1,2-дигидроксибензола и его производных до орто-хинона. 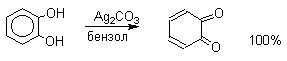 Для окисления фенолов, ароматических аминов и гидрохинонов до 1,4-бензохинонов и 1,4-нафтохинонов часто используют реагенты на основе хрома (YI). К ним относятся оксид хрома (YI) в уксусной кислоте или реагент Килиани (дихромат натрия в серной кислоте), однако выходы хинонов, как правило, ниже, чем при окислении солью Фреми или карбонатом серебра. 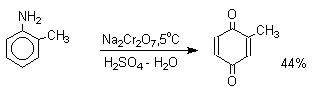 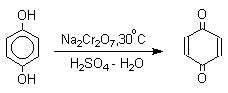 1,4-нафтохинон, 9,10-антрахинон и 9,10-фенантренхинон могут быть получены прямым окислением углеводородов оксидом хрома (YI) в уксусной кислоте или бихроматом натрия в серной кислоте. 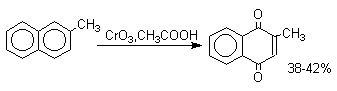 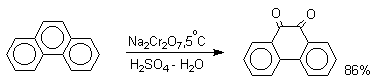 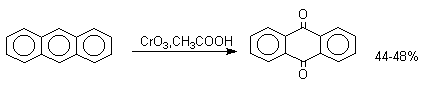 В промышленности тот же самый результат достигается при окислении кислородом в присутствии оксида ванадия (Y) как катализатора. Таким способом можно получать антрахинон и фенантренхинон. 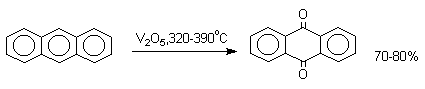 9,10-Антрахинон получают также ацилированием бензола фталевым ангидридом по Фриделю-Крафтсу с последующей циклизацией орто-бензоилбензойной кислоты. 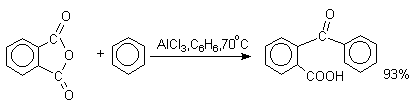 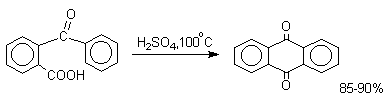 Этот один из самых простых и старых способов промышленного получения антрахинона. 3. Физические свойства и строение Хиноны – окрашенные кристаллические вещества. п-Бензохиноны окрашены в желтый цвет, о-бензохиноны – в красный, антрахиноны и фенантрахиноны – светло-желтые вещества. Карбонильные группы оказывают значительное электроноакцепторное влияние, поэтому хиноны обладают электроноакцепторными свойствами. Электроноакцепторные свойства хинонов характеризуются сродством к электрону и восстановительным потенциалом (см. ниже). Сродство к электрону возрастает при введении электроноакцепторных заместителей и уменьшается при увеличении числа конденсированных циклов. о-Хиноны более сильные акцепторы, чем п-хиноны. 4. Химические свойства Восстановление и образование семихинонов Наиболее важной реакцией хинонов является их восстановление до двухатомных фенолов. Восстановление хинонов осуществляется в две стадии. На первой стадии в результате одноэлектронного восстановления образуются анион-радикалы, которые называют семихинонами. Эти частицы могут быть легко зарегистрированы с помощью ЭПР-спекроскопии. На второй стадии анион-радикал присоединяет еще один электрон с образованием дианиона двухатомного фенола. 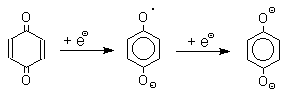 Способность хинона восстанавливаться до двухатомного фенола, т.е. свойство его как окислителя, оценивается с помощью нормального редокс-потенциала, определяемого из уравнения Нернста для реакции: 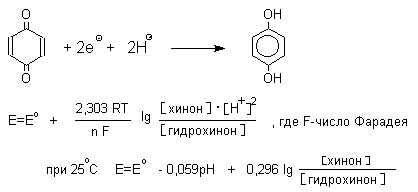 Величина Ео представляет собой нормальный потенциал, характерный для каждой системы хинон-гидрохинон, при равных концентрациях хинона и гидрохинона и концентрации ионов водорода, равной единице. Таким образом, Ео является количественной характеристикой окислительной способности хинона. Величины нормальных редокс-потенциалов Ео некоторых хинонов в воде при 25оС
Из данных, представленных в таблице следует, что 1,2-хиноны более сильные окислители, чем 1,4-хиноны, а бензохиноны превосходят по окислительной способности хиноны нафталинового ряда, которые в свою очередь превосходят антрахиноны и фенантренхиноны. Электроноакцепторные группы усиливают окислительные свойства хинонов. Высокие редокс-потенциалы хинонов определяются тем, что восстановление хинона в двухатомный фенол сопровождается превращением ненасыщенного кетона в ароматическое соединение. Восстановление хинонов до двухатомных фенолов осложняется образованием хингидрона - аддукта состава 1:1 между хиноном и двухатомным фенолом. Хингидрон может быть окислен до хинона или нацело восстановлен до гидрохинона. 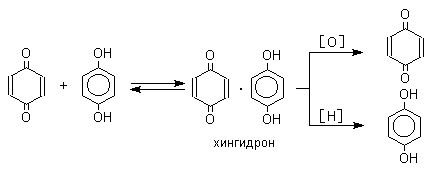 Окрашенный в темно-зеленый цвет хингидрон представляет собой классический пример молекулярных комплексов, где один компонент служит донором, а другой - является акцептором электрона. Такие комплексы, где происходит перекрывание ВЗМО донора и НСМО акцептора, получили название комплексов с переносом заряда. В кристаллах хингидрона молекулы хинона и гидрохинона чередуются и располагаются в двух параллельных плоскостяхдруг над другом. Комплексы с переносом заряда часто интенсивно окрашены. Окраска комплексов обусловлена переносом заряда от ароматического донора к акцептору, хотя степень переноса заряда невелика и редко превышает 0,1 заряда электрона. Восстановление хинонов до двухатомных фенолов проводят с помощью самых разнообразных восстановителей, среди которых в лабораторных условиях предпочтение отдается дитиониту натрия Na2S2O4 в щелочной среде. 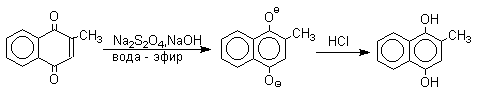 Помимо дитионита натрия в качестве восстановителей употребляются алюмогидрид лития и боргидрид натрия, хлорид олова (II) в соляной кислоте, цинк в уксусной кислоте и др. В промышленности восстановление 1,4-бензохинона до гидрохинона осуществляется с помощью оксида серы (IY)и железа в воде при 70-80оС. 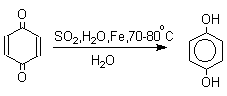 9,10-Антрахинон при восстановлении дитионитом натрия образует 9,10-антрадиол (антрагидрохинон). 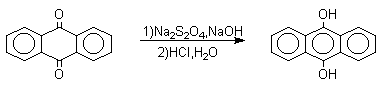 При восстановлении 9,10-антрахинона оловом в смеси соляной и уксусной кислот получается антрон - простейший кетон ряда антрацена. 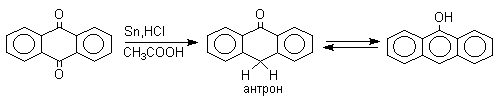 Производные 9,10-антрахинона широко используются в качестве синтетических антрахиноновых красителей для хлопка, шерсти и синтетических волокон. Эти красители отличаются яркостью цвета, высокой термической и фотохимической устойчивостью. Выше в качестве примера приведены формулы некоторых антрахиноновых красителей. Эти красители получают из 2-аминоантрахинона и его производных, например: 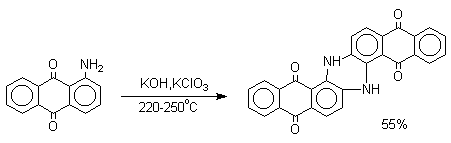 Восстановление антрахиноновых и других кубовых красителей дитионитом натрия в щелочной среде используется для перевода этих нерастворимых в воде соединений в так называемую лейкоформу, которая в виде динатриевой соли хорошо растворима в воде. Таким образом, например, упомянутый выше индантрен восстанавливают в тетрагидропроизводное, имеющее четыре фенольных гидроксила. Это лейкопроизводное хорошо растворимое в воде. Хлопчатобумажную ткань пропитывают раствором лейкоформы и выдерживают на воздухе. Лейкоформа окисляется кислородом до исходного красителя. Такой способ крашения гарантирует однородность окраски ткани. Легкость восстановления хинонов до фенола открывает возможность для использования хинонов в качестве дегидрирующих агентов. Для этой цели выбирают хиноны с высоким окислительно-восстановительным потенциалом, такие как 2,3,5,6-тетрахлор-1,4-бензохинон (хлоранил); 2,3-дихлор-5,6-дициано-1,4-бензохинон (ДДХ), дифенохинон. 1,2-Хиноны ввиду нестабильности практически не используются в качестве дегидрирующих агентов. Дегидрированию подвергаются дигидроароматические соединения ряда бензола и тетрагидропроизводные ряда нафталина, антрацена, гетероциклических соединений, тропилиден и т.д. 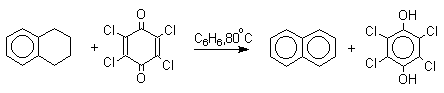 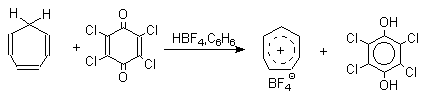 Механизм дегидрирования углеводородов заключается в отщеплении хиноном гидрид-иона с образованием карбокатиона, который стабилизируется отщеплением протона. Поэтому дегидрированию подвергаются углеводороды, которые при отщеплении гидрид-иона образуют сравнительно стабильные карбокатионы. Хиноны как a ,b -непредельные кетоны 1,4-Хиноны представляют собой типичныеa ,b -ненасыщенные кетоны и для них характерны реакции 1,2- и 1,4-присоединения к сопряженной системе. 1,4-Бензохинон присоединяет хлористый водород в 1,4-положение с образованием 2-хлоргидрохинона. 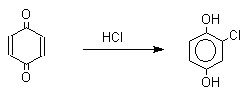 2-Хлоргидрохинон окисляется исходным хиноном до 2-хлор-1,4-бензохинона, который вновь присоединяет HCl с образованием 2,3-дихлоргидрохинона. 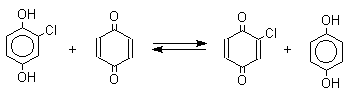 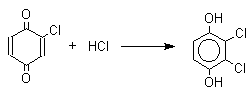 Для первичных аминов характерно сопряженное присоединение к 1,4-хинонам. При взаимодействии 1,4-бензохинона с анилином получается 2,5-бис(фениламино)-1,4-бензохинон. 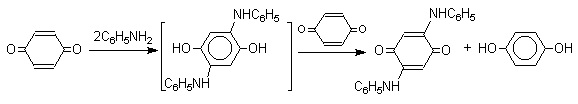 Аналогично происходит присоединение к 1,4-бензохинону и 1,4-нафтохинону спиртов, тиолов. Вместе с тем 1,4-хиноны вступают в типичные реакции 1,2-присоединения по карбонильной группе и с гидроксиламином дают моно- и диоксимы. 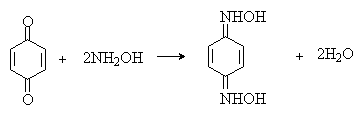 Хиноны как диенофилы в реакции диенового синтеза 1,4-Бензохинон, 1,4-нафтохинон и их производные проявляют свойства активных диенофилов в реакции Дильса-Альдера. При взаимодействии 1,3-бутадиена с 1,4-бензохиноном при 25оС получается моноаддукт, который медленно енолизуется с образованием соответствующего гидрохинона. Это превращение, как и следовало ожидать, катализируется кислотой. При последующем окислении оксидом хрома (YI) получается 1,4-нафтохинон. 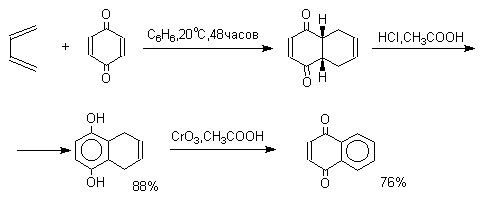 Электроноакцепторные заместители в хиноне активируют диенофил, а электронодонорные заместители замедляют присоединение 1,3- диенов. ДДХ и 1,2,3,5-тетрациан-1,4-бензохинон исключительно эффективны в качестве диенофилов. Диеновый синтез с участием 1,4-бензохинона используется для получения полициклических конденсированных ароматических углеводородов. 1 2 |