1. о сущности живого. Нуклеопротеидные комплексы. Эволюция представлений о химической сущности жизни. Ф. Энгельс Жизнь способ существования белковых тел
 Скачать 17.11 Mb. Скачать 17.11 Mb.
|

|
85. Роль ферментов в клеточном метаболизме. Энзимопатии. В основебольшинства заболеванийи патологических состояний лежит нарушение функционирования ферментов в клетке –энзимопатии. - первичные (наследственные) обычно относят к метаболическим болезням. - вторичные (преобретённые) энзимопатии. нарушение образования конечного продукта. Например, а) приальбинизмеимеет место недостаточность ферментатирозингидроксилазы(тирозиназы), что нарушает синтез меланина. Наблюдается слабая пигментация кожи, волос, красноватый цвет радужки. б) недостаток катехоламинов припаркинсонизме. · накопление субстратов предшественников. Имеет место приалкаптонурии– нарушении активности фермента окисления гомогентизиновой кислоты –диоксигеназы гомогентизиновой кислоты. Кроме указанных, распространенными первичными энзимопатиями являютсягалактоземия, недостаточностьлактазыисахаразы. Вторичные(приобретенные) энзимопатии возникают как следствие заболеваний органов, вирусных инфекций и т.п., что приводит к нарушению синтеза фермента или условий его работы, примером может служить недостаточность ферментов желудочно-кишечного тракта при заболеваниях желудка, поджелудочной железы или желчного пузыря. Недостаток витаминов и их коферментных форм также является причиной приобретенных ферментопатий. 1. Метаболизм — совокупность химических реакций в клетке: расщепления (энергетический обмен) и синтеза (пластический обмен). 2. Функции клеточного обмена веществ: 1) обеспечение клетки строительным материалом, необходимым для образования клеточных структур; 2) снабжение клетки энергией, которая используется на процессы жизнедеятельности (синтез веществ, их транспорт и др.). 3. Энергетический обмен — окисление органических веществ (углеводов, жиров, белков) и синтез богатых энергией молекул АТФ за счет освобождаемой энергии. 4. Пластический обмен — синтез молекул белков из аминокислот, полисахаридов из моносахаридов, жиров из глицерина и жирных кислот, нуклеиновых кислот из нуклеотидов, использование на эти реакции энергии, освобождаемой в процессе энергетического обмена. 5. Ферментативный характер реакций обмена. Ферменты — биологические катализаторы, ускоряющие реакции обмена в клетке. Ферменты — в основном белки, у некоторых из них есть небелковая часть (например, витамины). Молекулы ферментов значительно превышают размеры молекул вещества, на которые они действуют. Активный центр фермента, его соответствие структуре молекулы вещества, на которое он действует. 6. Разнообразие ферментов, их локализация в определенном порядке на мембранах клетки и в цитоплазме. Подобная локализация обеспечивает последовательность реакций. 7. Высокая активность и специфичность действия ферментов: ускорение в сотни и тысячи раз каждым ферментом одной или группы сходных реакций. Условия действия ферментов: определенная температура, реакция среды (рН), концентрация солей. Изменение условий среды, например рН, — причина нарушения структуры фермента, снижения его активности, прекращения действия. 86. Генетическая детерминация структуры гемоглобина. Гемоглобинопатии. Молекулы гемоглобина- тетрамеры , содержащие по 2 пары гемоглобиновых цепей различного типа. В ходе развития организма наблюдают образование различных вариантов Нв в зависимости от экспрессии разных глобиновых типов. Различают: 1. Эмбриональный Нb - состоит из 2ζ(дзета) цепей и 2 ε(эпсилон) 2. Фетальный Нb – состоит из 2α(альфа) и 2γ (гамма) 3. Нb взрослых – состоит из 2α(альфа) и 2 β(бетта) В течении 2 месяца беременности снижается синтез ζ и ε цепей, а усиливается синтез α и γ.- образуется фетальный гемоглобин. На 3 месяце беременности активируются гены β и δ (дельта) цепей глобина, тогда как концентрация γ падает, это переключение ускоряется и фетальный гемоглобин заменяется на взрослый. Преобладающим типом гемоглобина является гемоглобинА (α2β2), α и β цепи различаются по многим аминокислотным остаткам. У всех взрослых помимо Нв-А, еще и Нв-А2 (α2δ2), характерная для А2 δ цепь отличается от β цепи только по 10 аминокислотным остаткам. Около 1% фетального Нв может присутствовать и во взрослом организме. γ цепь значительно отличается от α и β и обладает бОльшим сродством к кислороду, так как плод получает кислород через плаценту(но там содержание не высокое), поэтому фетальный Нв содержит γ цепь. Известно 2 типа γ цепей:
Ζ цепи похожи по аминокислотному составу на α , а ε цепей на β. Синтез γ цепей у эмбриона происходит в печени, селезенке и костном мозге. Синтез β цепей происходит в костном мозге. Все глобиновые цепи имеют общее эволюционное развитие, возникают в последствии дупликации генов и их дальнейшей модификации. Синтез небелковых гемогрупп также кодируется генами(т к они кодируют структуру ферментов обеспечивающих биосинтез гемма). Глобиновые гены распологаются в 11 и 16 хромосомах. И образуют α и β подобные кластеры. Α подобные кластеры располагаются в коротком плече 16 хромосомы, а β подобные кластеры в коротком плече 11 хромосомы Структурные гены расположены в порядке от 5’ к 3’ концу. Все глобиновые гены имеют по 3 экзона(информативные участки) и 2 интрона (неинформативные участки),интроны транскрибируются вместе с экзонами, поэтому они есть в первичном транскрипте. Процессинг- созревание первичного транскрипта( интрены вырезаются, экзона сшиваются). Гемоглобинопатии - это группа патологических состояний, обусловленные нарушениями структуры цепей глобина (заменой одной или нескольких аминокислот в цепи глобина, отсутствие участка цепи или ее удлинением.) Существуют четыре основных типа болезней гемоглобина: 1. Гемолитические анемии, вызванные нестабильностью гемоглобина. 2. Метгемоглобинемии, обусловленные ускоренным окислением гемоглобина. 3. Эритроцитоз, вызванный нарушением сродства гемоглобина к кислороду. 4. Серповидно-клеточные нарушения как следствие повреждений клеточных мембран эритроцитов. Гемолитические анемии. Они вызываются нестабильными формами гемоглобина. В большинстве случаев мутация затрагивает β-цепь. У многих нестабильных гемоглобинов в полипептидной цепи обнаруживаются аминокислотные замены или делеции в участках связывания гема. Нестабильность может быть едва заметной, что не имеет никаких клинических последствий, до выраженной нестабильности, при которой происходит интенсивное разрушение эритроцитов. Нестабильность часто обусловлена преждевременной диссоциацией гема и глобиновых цепей. Точный диагноз может быть затруднен, особенно если не наблюдается изменений электрофоретической подвижности. В таком случае необходимо выделение глобиновых цепей для дальнейшего анализа в специализированных лабораториях. Метгемоглобинемия, обусловлена ускоренным окислением двухвалентного железа до трехвалентного. Больные с мутацией в α–цепи, вызывающими образование HbМ, страдают цианозом от рождения. При мутации в β-цепи цианоз развивается только через 6 месяцев после рождения, когда происходи замена γ–цепи на β-цепь. Эритроцитоз, вызванный образованием гемоглобинов с нарушенным сродством к кислороду. Существует около 30 гемоглобинов с повышенным сродством к кислороду. Повышенное сродство к кислороду приводит к уменьшению количества кислорода, освобождающегося из комплекса с гемом в тканях организма, и вызывает гипоксию. Гипоксия ведет к выделению гормона эритропоэтина, стимулирующего образование эритроцитов и собственно эритроцитоз. Было обнаружено всего три гемоглобина с уменьшенным сродством к кислороду. При таком дефекте количество кислорода, поступающее в ткани, увеличивается, поэтому следует ожидать уменьшение синтеза эритропоэтина. В двух случаях, как и следовало ожидать, наблюдалась слабовыраженная анемия. Серповидно-клеточная анемия — это наследственная гемоглобинопатия, связанная с таким нарушением строения белка гемоглобина, при котором он приобретает особое кристаллическое строение — так называемый гемоглобин S. Эритроциты, несущие гемоглобин S вместо нормального гемоглобина А, под микроскопом имеют характерную серпообразную форму (форму серпа). Эритроциты, несущие гемоглобин S, обладают пониженной стойкостью и пониженной кислород-транспортирующей способностью, поэтому у больных с серповидноклеточной анемией повышено разрушение эритроцитов в селезенке, укорочен срок их жизни, повышен гемолиз и часто имеются признаки хронической гипоксии (кислородной недостаточности) или хронического «перераздражения» эритроцитарного ростка костного мозга. Больные серповидноклеточной анемией обладают повышенной (хотя и не абсолютной) врождённой устойчивостью к заражению различными штаммами малярийного плазмодия. Симптомы серповидноклеточной анемии делятся на две основные категории. Из-за хрупкости красных клеток крови всегда наблюдается анемия, которая может привести к потере сознания, делает больного физически менее выносливым и может вызвать желтуху (связанную с чрезмерным распадом гемоглобина). 87. Задачи медико-генетического консультирования. Медико-генетическое консультирование-отрасль профилактической медицины, главной целью которой является предупреждение рождения детей с наследственной патологией. Современная генетическая консультация призвана служить интересам семьи и общества. Цель: установления степени генетического риска в обследуемой семье и разъяснение супругам в доступной форме медико-генетического заключения. Задачи медико-генетического консультирования: 1.про- и ретроспективное (до и после рождения) консультирование семей с наследственной или врожденной патологией 2.пренатальная диагностика врожденных и наследственных заболеваний 3.помощь врачам различных специальностей в постановке диагноза заболевания, если для этого требуются специальные генетические методы исследования 4.объяснение в доступной форме пациенту и его семье степени риска иметь больных детей и помощь им в принятии решения 5.ведение территориального регистра семей и больных с наследственной и врожденной патологией и их диспансерное наблюдение 6.пропаганда медико-генетических знаний среди населения. Коротко говоря, задачей генетической консультации является составление генетического прогноза в семье индивидуума с аномалией физического, психического либо полового развития и выбор профилактических мероприятий по предупреждению рождения больного ребенка. 88. Человек как специфический объект генетического анализа. Медико-генетическое консультирование и прогнозирование. Медико-генетическое консультирование-отрасль профилактической медицины, главной целью которой является предупреждение рождения детей с наследственной патологией. Современная генетическая консультация призвана служить интересам семьи и общества. Цель: установления степени генетического риска в обследуемой семье и разъяснение супругам в доступной форме медико-генетического заключения. Показаниями для медико-генетического консультирования являются: 1) рождение ребенка с врожденным пороком развития 2) установленная или подозреваемая наследственная болезнь в семье в широком смысле слова 3) задержка физического развития или умственная отсталость у ребенка 4) повторные спонтанные аборты, выкидыши, мертворождения 5) близкородственные браки 6) неблагополучное протекание беременности. В принципе каждая супружеская пара должна пройти медико- генетическое консультирование до планирования деторождения (проспективно) и безусловно после рождения больного ребенка (ретроспективно). Задачи медико-генетического консультирования: 1.про- и ретроспективное (до и после рождения) консультирование семей с наследственной или врожденной патологией 2.пренатальная диагностика врожденных и наследственных заболеваний 3.помощь врачам различных специальностей в постановке диагноза заболевания, если для этого требуются специальные генетические методы исследования 4.объяснение в доступной форме пациенту и его семье степени риска иметь больных детей и помощь им в принятии решения 5.ведение территориального регистра семей и больных с наследственной и врожденной патологией и их диспансерное наблюдение 6.пропаганда медико-генетических знаний среди населения. Коротко говоря, задачей генетической консультации является составление генетического прогноза в семье индивидуума с аномалией физического, психического либо полового развития и выбор профилактических мероприятий по предупреждению рождения больного ребенка. Первый этап медико-генетического консультирования заключается во всестороннем обследовании больного, направленном на уточнение диагноза. Значительное внимание на этом этапе уделяется сбору генеалогической информации и анамнестических данных. Предположение о наследственном характере заболевания и установление типа его наследования может быть сделано на основании анализа родословной пробанда и тщательном обследовании всех членов консультирующейся семьи. 89. Мутации, несовместимые с жизнью человека. Любое изменение последовательности ДНК называется мутацией. Мутации подразделяются на геномные, хромосомные и генные. Мутации можно классифицировать по их влиянию на жизнеспособность: - витальные мутации не оказывают влияние на жизнеспособность - супервитальные они способствуют повышению жизнеспособности, примером является гетерозоготы по серповидноклеточной анемии - полулетальные мутации снижение жизнеспособности, например альбинизм, синдром Дауна - летальные мутации несовместимы с жизнью синдром Патау, синдром Эдвардца. Геномные мутации обусловлены изменением числа хромосом (увеличением или уменьшением). Большинство геномных мутаций несовместимо с жизнью. Эмбрионы и плоды погибают и удаляются из организма матери в ранние сроки беременности (самопроизвольный выкидыш). Наиболее частая форма геномных мутаций, совместимых с жизнью, –– синдромы трисомий. Они являются, как правило, следствием нерасхождения хромосом в мейозе и проявляются множественными врожденными пороками развития. Примером является синдром трисомии по 21–й хромосоме, известный как синдром Дауна. Такие мутации не ведут к изменению жизнеспособности и называются сейнсис мутации. Может измениться первичная структура белка на одну аминокислоту (спецефичность генетического кода). Последствия такой замены различны, т.е. это либо миссенс мутации, когда белок функционирует плохо. Первичная структура белка может стать короче из-за появления нонсенс кодона, нонсенс мутации, прекращается синтез белка. Белок может удлиниться, если замена произойдет в нонсенс кодане. Белок может стать короче из-за изменения кадона инициации. Синдром Патау - хромосомная аномалия, представляющая собой трисомию по 13-ой паре аутосом. При синдроме Патау у ребенка имеются множественные и крайне тяжелые аномалии развития, определяющие частые случаи внутриутробной гибели плода и малую продолжительность жизни детей с данной патологией. Причины - присутствие в кариотипе дополнительной копии 13-ой хромосомы. В большинстве случаев (75-80%) имеет место простая полная трисомия, связанная с нерасхождением 13-ой хромосомы в мейозе у одного из родителей (чаще у матери). При этом все без исключения клетки плода имеют кариотип 47,XX 13+ или 47,XY 13+. Меньшая часть случаев синдрома Патау представлена несбалансированными транслокациями хромосом 13-й пары, мозаичными формами, изохромосомой. Генетический сбой может произойти во время формирования гамет или уже на этапе образования зиготы. Генетическая мутация в гаметогенезе или зародышевой клетке в основном возникает de novo, как случайное событие. Наследственные формы синдрома Патау связаны с наличием робертсоновской (сбалансированной) транслокации у родителей. Вновь возникшая робертсоновская транслокация может наследоваться, не вызывая синдром Патау у ребенка, но увеличивая риск рождения детей с данной аномалией в последующих поколениях. Синдром Эдвардса (синдром трисомии 18) — второе по частоте после болезни Дауна хромосомное заболевание, хар комплексом множественных пороков развития и трисомией 18 хромосомы. Девочки болеют в 3 раза чаще, чем мальчики. Причины - стабилизирующем действие Х-хромосомы при аберрации 18 пары, тогда как зиготы с трисомией 18, имеющие мужской генотип, элиминируются. - оплодотворения яйцеклетки с лишней хромосомой 18 сперматозоидом, имеет Х-хромосому. Продолжительность беременности превышает нормальную (в среднем 42 недели), диагностируют слабую активность плода, многоводие, плацента малых размеров, часто оказывается только одна пупочная артерия; часть детей рождается в состоянии асфиксии, с очень низкой массой тела и резкой гипотрофией. Во время цитогенетического обследования в 80% случаев выявляют трисомия 18, а у 10% больных - мозаицизм. Описаны случаи транслокационного варианта, двойной анеуплоидии типа 48, XXV +18 с участием трисомного за хромосомы 18 клона. 90. Изменение геномной организации наследственного материала. Геномные мутации. Число, форма и размер хромосом являются систематическими признаками для каждого вида. В некоторых случаях при нарушении механизмов митоза или мейоза происходит нарушение процесса расхождения хромосом к полюсам клетки, приводящее к нерасхождению хромосом, а также к процессу удвоения хромосом. В результате тех и других нарушений возникают клетки с измененным числом хромосом Изменение числа хромосом может происходить за счет увеличения или уменьшения числа целых гаплоидных наборов или отдельных хромосом. Организмы, у которых произошло умножение целых гаплоидных наборов, называют полиплоидными. Организмы, у которых число хромосом не является кратным гаплоидному, называют анеуплоидными. Возникновение геномных мутаций у млекопитающих известно только в качестве аномалий и приводит к гибели эмбриона на ранних стадиях развития. Однако изредка вследствие нерасхождения половых хромосом либо при одном, либо при двух делениях мейоза, возникает так называемая трисомия, а также моносомия-ХО, вызывающая серьезные гормональные нарушения у плода. Синдром Шерешевского-Тернера (45 хромосом, половые хромосомы – ХО). Частота встречаемости заболевания 1:2500. Заболевание сопровождается характерными аномалиями физического развития, низкорослостью, кожными крыловидными складками на боковых поверхностях шеи, деформацией локтевых суставов и недоразвитием вторичных половых признаков Синдром Клайнфельтера (47 хромосом, половые хромосомы – ХХУ). Частота встречаемости заболевания 1:1100. Для мужчин с синдромом Клайнфельтера характерны высокий рост, длинные конечности, бесплодие, повышенное выделение женских половых гормонов, склонность к ожирению. Лишняя Х хромосома обусловливает различные нарушения психики. Больные очень внушаемы, вялы, апатичны, безынициативны, у них часто отмечается умственная отсталость. Клиническая картина начинает проявляться у мальчиков в период полового созревания 91. Причины гетероплоидии у человека Гетероплоидия (анеуплоидия)-некратное увеличение или уменьшение числа хромосом. Чаще всего наблюдается уменьшение или увеличение числа хромосом на одну (реже две и более). Наиболее вероятной причиной гетероплоидии является нерасхождение какой-либо пары гомологичных хромосом во время мейоза у кого-то из родителей. В этом случае одна из образовавшихся гамет содержит на одну хромосому меньше, а другая — на одну больше. Слияние таких гамет с нормальной гаплоидной гаметой при оплодотворении приводит к образованию зиготы с меньшим или большим числом хромосом по сравнению с диплоидным набором, характерным для данного вида: нулесомия (2n - 2), моносомия (2n - 1), трисомия (2n + 1), тетрасомия (2n + 2) и т.д. По типу вовлечённых хромосом выделяют анеуплоидию половых хромосом и аутосомную анеуплоидию. Анеуплоидия по половым хромосомам характеризуется значительно более мягкими фенотипическими проявлениями, чем анеуплоидия по аутосомам. 92. Изменения нуклеотидных последовательностей ДНК. Генные мутации Генные (точковые) мутации - это изменения числа и/или последовательности нуклеотидов в структуре ДНК (вставки, выпадения, перемещения, замещения нуклеотидов) в пределах отдельных генов, приводящие к изменению количества или качества соответствующих белковых продуктов. Замены оснований приводят к появлению трех типов мутантных кодонов: -с измененным смыслом (миссенс-мутации) -с неизмененным смыслом (нейтральные мутации) -бессмысленных или терминирующих кодонов (нонсенс-мутации). В результате миссенс-мутании в кодируемом данным геном полипептиде одна аминокислота замещается на другую, поэтому фенотипическое проявление мутации зависит от функциональной значимости затронутого домена. Так замены аминокислот в активных центрах белков могут сопровождаться полной потерей их функциональной активности. Не всякая замена аминокислоты отразится на функциональной активности белка, вследствие чего происшедшая мутация может остаться не выявленной. Этим объясняется факт отмечаемого несовпадения частоты мутаций в определенном гене и встречаемости мутантов по нему. Кроме того, в силу вырожденности генетического кода, не всякая замена основания приведет к миссенс-мутации, возможно, она окажется нейтральной. В результате нонсенс мутации кодон, определяющий какую-либо аминокислоту, превращается в один из стоп-кодонов, не транслирующихся на рибосомах (УAA, УAГ, УГA). Появление такого кодона не в конце структурного гена, а внутри него, приводит к преждевременной терминации трансляции и обрыву полипептидной цепи. Нонсенс-мутации обладают наибольшим повреждающим действием, так как образующиеся при преждевременной терминации трансляции белки не способны к модификации, часто не защищены от действия протеолитических ферментов и быстро деградируют. Вставки, перемещения или выпадения отдельных оснований или их коротких последовательностей в пределах гена вызывают сдвиг рамки считывания. 93. Изменение структурной организации хромосом. Хромосомные мутации. Хромосомные мутации— изменения положения участков хромосом; приводят к изменению размеров и формы хромосом. В эти изменения могут быть вовлечены как участки одной хромосомы, так и участки разных, негомологичных хромосом, поэтому хромосомные мутации (перестройки) подразделяются на внутри- и межхромосомные. А.внутрихромосомные мутации 1.хромосомные дупликации-удвоение участка хромосомы. 2.хромосомные делеции-утрата хромосомой какого-либо участка. 3.хромосомные инверсии-разрыв хромосомы, переворачивание оторвавшегося участка на 180° и встраивание его на прежнее место. Б.межхромосомные мутации 1.транслокация-обмен участками между негомологичными хромосомами (в мейозе). 2.транспозиция-включение участка хромосомы в другую, негомологичную хромосому без взаимного обмена. 94. Методы в генетике человека. Генеалогический метод. Принципы построения родословных и их типы. В основе этого метода лежит составление и анализ родословных. Этот метод широко применяют с древних времен и до наших дней в коневодстве, селекции ценных линий крупного рогатого скота и свиней, при получении чистопородных собак, а также при выведении новых пород пушных животных. Родословные человека составлялись на протяжении многих столетий в отношении царствующих семейств в Европе и Азии. Как метод изучения генетики человека генеалогический метод стали применять только с начала XX столетия, когда выяснилось, что анализ родословных, в которых прослеживается передача из поколения в поколение какого-то признака (заболевания), может заменить собой фактически неприменимый в отношении человека гибридологический метод. Сбор сведений о семье начинается с человека, называемого пробандом. По завершении сбора сведений приступают к географическому изображению родословной. Для этого используют условные обозначения, предложенные Г. Юстом в 1931 г. (рис. 6.24). П 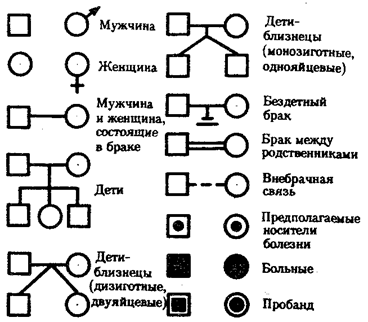 околения в родословной обозначаются римскими цифрами, которые ставятся слева от родословной. Арабскими цифрами нумеруются родственники одного поколения слева направо. Братья и сёстры располагаются в порядке рождения и именуются сибсами. Изображение родословной необходимо начинать с самого старшего поколения. К родословной записывается легенда. околения в родословной обозначаются римскими цифрами, которые ставятся слева от родословной. Арабскими цифрами нумеруются родственники одного поколения слева направо. Братья и сёстры располагаются в порядке рождения и именуются сибсами. Изображение родословной необходимо начинать с самого старшего поколения. К родословной записывается легенда.Анализ родословной включает следующие этапы: 1. Установление, является ли данный признак или заболевание единичным в семье или имеется несколько случаев (семейный характер). Если признак встречается несколько раз в разных поколениях, то можно предположить, что он имеет наследственную природу. 2. Определение типа наследования признака. Для этого анализируют родословную, учитывая следующие моменты: 1) встречается ли изучаемый признак во всех поколениях и многие ли члены родословной обладают этим признаком; 2) одинакова ли частота признака у лиц обоих полов, у лиц какого пола он встречается чаще; 3) лицам какого пола передается признак от больного отца и больной матери; 4) есть ли в родословной семьи, в которых у обоих здоровых родителей рождались больные дети или у обоих больных родителей рождались здоровые дети; 5) какая часть потомства в семьях, где болен один из родителей, имеет наследуемый признак. Аутосомно-доминантное наследование: 1) признак проявляется в равной степени у представителей обоих полов; 2) признак встречается в каждом поколении; 3) дети, имеющие признак, рождаются в семье, где хотя бы один из родителей имеет этот признак Аутосомно-рецессивное наследование: 1) признак проявляется в равной степени у представителей обоих полов; 2) признак встречается не в каждом поколении, число людей, имеющих признак, относительно; 3) дети, имеющие признак, обычно рождаются в семьях, где оба родителя этого признака не имеют; частота рождения детей с признаком возрастает в родственных браках. Х - сцепленное доминантное наследование 1) от женщины признак может передоваться как сыновьям, так и дочерям; 2) от мужчины признак наследуют только его дочери, все его сыновья признака не имеют Х - сцепленное рецессивное наследование 1) признак, как правило, проявляется только у мужчин; 2) гетерозиготные женщины-носительницы признака не имеют, но у них могут быть потомки мужского пола с этим признаком У - сцепленное наследование 1) признак имеют только мужчины; 2) признак передаётся только по мужской линии Цитоплазматическое наследование 1) признак передаётся от матери всем детям независимо от пола; 2) отцы, имеющие признак, не передают его ни сыновьям, ни дочерям 95. Методы в генетике человека. Цитогенетический метод. Кариотип человека. Цитогенетический метод основан на микроскопическом изучении хромосом в клетках человека. Его стали широко применять в исследованиях генетики человека с 1956 г., когда шведские ученые Дж. Тийо и А. Леван, предложив новую методику изучения хромосом, установили, что в кариотипе человека 46, а не 48 хромосом, как считали ранее. Современный этап в применении цитогенетического метода связан с разработанным в 1969 г. Т. Касперсоном методом дифференциального окрашивания хромосом, который расширил возможности цитогенетического анализа, позволив точно идентифицировать хромосомы по характеру распределения в них окрашиваемых сегментов (см. разд. 3.5.2.3). Применение цитогенетического метода позволяет не только изучать нормальную морфологию хромосом и кариотипа в целом, определять генетический пол организма, но, главное, диагностировать различные хромосомные болезни, связанные с изменением числа хромосом или с нарушением их структуры. Кроме того, этот метод позволяет изучать процессы мутагенеза на уровне хромосом и кариотипа. Применение его в медико-генетическом консультировании для целей пренатальной диагностики хромосомных болезней дает возможность путем своевременного прерывания беременности предупредить появление потомства с грубыми нарушениями развития. Материалом для цитогенетических исследований служат клетки человека, получаемые из разных тканей — лимфоциты периферической крови, клетки костного мозга, фибробласты, клетки опухолей и эмбриональных тканей и др. Непременным требованием для изучения хромосом является наличие делящихся клеток. Непосредственное получение таких клеток из организма затруднено, поэтому чаще используют легкодоступный материал, каковым являются лимфоциты периферической крови. В норме эти клетки не делятся, однако специальная обработка их культуры фитогемагглютинином возвращает их в митотический цикл. Накопление делящихся клеток в стадии метафазы, когда хромосомы максимально спирализованы и хорошо видны в микроскоп, достигается обработкой культуры колхицином или колцемидом, разрушающим веретено деления и препятствующим расхождению хроматид. Микроскопирование мазков, приготовленных из культуры таких клеток, позволяет визуально наблюдать хромосомы. Фотографирование метафазных пластинок и последующая обработка фотографий с составлением кариограмм, в которых хромосомы выстроены парами и распределены по группам, позволяют установить общее число хромосом и обнаружить изменения их количества и структуры в отдельных парах. В качестве экспресс-метода, выявляющего изменение числа половых хромосом, используют метод определения полового хроматина в неделящихся клетках слизистой оболочки щеки. Половой хроматин, или тельце Барра, образуется в клетках женского организма одной из двух Х-хромосом. Оно выглядит как интенсивно окрашенная глыбка, расположенная у ядерной оболочки (см. рис. 3.77). При увеличении количества Х-хромосом в кариотипе организма в его клетках образуются тельца Барра в количестве на единицу меньше числа Х-хромосом. При уменьшении числа Х-хромосом (моносомия X) тельце Барра отсутствует. В мужском кариотипе Y-хромосома может быть обнаружена по более интенсивной по сравнению с другими хромосомами люминесценции при обработке их акрихинипритом и изучении в ультрафиолетовом свете. 96. Кариотип человека. Денверская и Парижская классификация хромосом. Кариотип человека (от греч. - орех, ядро и - отпечаток, тип) — диплоидный хромосомный набор человека, представляющий собой совокупность морфологически обособленных хромосом, внесённых родителями при оплодотворении. Хромосомы набора генетически неравноценны: каждая хромосома содержит группу разных генов. Все хромосомы в кариотипе человека делятся на аутосомы и половые хромосомы. В кариотипе человека 44 аутосомы (двойной набор) - 22 пары гомологичных хромосом и одна пара половых хромосом — XX у женщин и ХУ у мужчин. Денверская классификация хромосом 22 пары аутосом разделены на семь групп, обозначаемых буквами от А до G. Каждая группа хромосом характеризуется следующими особенностями: Группа А содержит 3 пары длинных хромосом (1-3), каждую из которых можно легко индивидуализировать. Метацентрические (центромера располагается в середине длины хромосомы) Группа В содержит две пары хромосом (4-5). Они короче хромосом из группы А и являются субметацентрическими (центромера располагается ближе к одному концу); Группа С содержит 6 пар аутосом (6-12), все хромосомы с субмедиально расположенной центромерой, средних размеров, их трудно индивидуализировать. К этой группе по размеру относится Х-хромосома, которая отличается тем, что заканчивает синтез ДНК позднее других Группа D содержит 3 пары хромосом (13-15). Хромосомы средних размеров, акроцентрические (центромера располагается на теломерном конце). Группа Е состоит из 3 пар коротких хромосом (16-18). Хромосомы 16-й пары являются метацентрическими (центромера в середине длины хромосомы). Хромосомы 17-й и 18-й пары, похожи между собой, отличаются меньшей общей длиной и размерами коротких плеч, являются субметацентрическими. Группа F имеет 2 пары коротких метацентрических хромосом (19-20), которые неотличимы друг от друга; Группа G состоит из 2-х пар хромосом (21-22). Это очень короткие акроцентрические хромосомы со спутниками, трудно различимы, хотя несколько отличаются по величине и морфологии. К ним примыкает У-хромосома, которая несколько длиннее и имеет на длинном плече вторичную перетяжку. 97. Методы в генетике человека. Близнецовый метод. Этот метод заключается в изучении закономерностей наследования признаков в парах одно- и двуяйцевых близнецов. Он предложен в 1875 г. Гальтоном первоначально для оценки роли наследственности и среды в развитии психических свойств человека. В настоящее время этот метод широко применяют в изучении наследственности и изменчивости у человека для определения соотносительной роли наследственности и среды в формировании различных признаков, как нормальных, так и патологических. Он позволяет выявить наследственный характер признака, определить пенетрантность аллеля, оценить эффективность действия на организм некоторых внешних факторов (лекарственных препаратов, обучения, воспитания). Суть метода заключается в сравнении проявления признака в разных группах близнецов при учете сходства или различия их генотипов. Монозиготные близнецы, развивающиеся из одной оплодотворенной яйцеклетки, генетически идентичны, так как имеют 100% общих генов. Поэтому среди монозиготных близнецов наблюдается высокий процент конкордантных пар, в которых признак развивается у обоих близнецов. Сравнение монозиготных близнецов, воспитывающихся в разных условиях постэмбрионального периода, позволяет выявить признаки, в формировании которых существенная роль принадлежит факторам среды. По этим признакам между близнецами наблюдается дискордантность, т.е. различия. Напротив, сохранение сходства между близнецами, несмотря на различия условий их существования, свидетельствует о наследственной обусловленности признака. Сопоставление парной конкордантности по данному признаку у генетически идентичных монозиготных и дизиготных близнецов, которые имеют в среднем около 50% общих генов, дает возможность более объективно судить о роли генотипа в формировании признака. Высокая конкордантность в парах монозиготных близнецов и существенно более низкая конкордантность в парах дизиготных близнецов свидетельствуют о значении наследственных различий в этих парах для определения признака. Сходство показателя конкордантности у моно- и дизиготных близнецов свидетельствует о незначительной роли генетических различий и определяющей роли среды в формировании признака или развития заболевания. Достоверно различающиеся, но достаточно низкие показатели конкордантности в обеих группах близнецов дают возможность судить о наследственной предрасположенности к формированию признака, развивающегося под действием факторов среды. Установление соотносительной роли наследственности и среды в развитии различных патологических состояний позволяет врачу правильно оценить ситуацию и проводить профилактические мероприятия при наследственной предрасположенности к заболеванию или осуществлять вспомогательную терапию при его наследственной обусловленности. Трудности близнецового метода связаны, во-первых, с относительно низкой частотой рождения близнецов в популяции (1:86—1:88), что осложняет подбор достаточного количества пар с данным признаком; во-вторых, с идентификацией монозиготности близнецов, что имеет большое значение для получения достоверных выводов. Для идентификации монозиготности близнецов применяют ряд методов. 1. Полисимптомный метод сравнения близнецов по многим морфологическим признакам (пигментации глаз, волос, кожи, форме волос и особенностям волосяного покрова на голове и теле, форме ушей, носа, губ, ногтей, тела, пальцевым узорам). 2. Методы, основанные на иммунологической идентичности близнецов по эритроцитарным антигенам (системы АВО, MN, резусу), по сывороточным белкам (γ-глобулину). 3. Наиболее достоверный критерий монозиготности предоставляет трансплантационный тест с применением перекрестной пересадки кожи близнецов. Несмотря на трудоемкость близнецового метода и возможность ошибок при определении монозиготности близнецов, высокая объективность выводов делает его одним из широко применяемых методов генетических исследований у человека. 98. Методы в генетике человека. Биохимический метод. Дерматоглифика. Биохимические методы изучают наследственные заболевания, обусловленные генными мутациями, а также полиморфизм по нормальным первичным продуктам генов. Впервые эти методы стали применять для диагностики генных болезней еще в начале XX в. В последние 30 лет их широко используют в поиске новых форм мутантных аллелей. С их помощью описано более 1000 врожденных болезней обмена веществ. Наиболее распространенными среди таких заболеваний являются болезни, связанные с дефектностью ферментов, структурных, транспортных или иных белков. Дефекты структурных и циркулирующих белков выявляются при изучении их строения. Установлено большое разнообразие гемоглобинов у человека, связанное с изменением структуры его пептидных цепей, что нередко является причиной развития заболеваний. Дефекты ферментов устанавливают путем определения содержания в крови и моче продуктов метаболизма, являющихся результатом функционирования данного белка. Дефицит конечного продукта, сопровождающийся накоплением промежуточных и побочных продуктов нарушенного метаболизма, свидетельствует о дефекте фермента или его дефиците в организме. Биохимическую диагностику наследственных нарушений обмена проводят в два этапа. На первом этапе отбирают предположительные случаи заболеваний, на втором — более точными и сложными методами уточняют диагноз заболевания. Применение биохимических исследований для диагностики заболеваний в пренатальном периоде или непосредственно после рождения позволяет своевременно выявить патологию и начать специфические медицинские мероприятия, как, например, в случае фенилкетонурии. Для определения содержания в крови, моче или амниотической жидкости промежуточных, побочных и конечных продуктов обмена кроме качественных реакций со специфическими реактивами на определенные вещества используют хроматографические методы исследования аминокислот и других соединений. Все многообразие биохимических методов делится на две группы: а) Методы, основанные на выявлении определенных биохимических продуктов, обусловленных действием разных аллелей. Легче всего выявлять аллели по изменению активности ферментов или по изменению какого-либо биохимического признака. б) Методы, основанные на непосредственном выявлении измененных нуклеиновых кислот и белков с помощью гель-электрофореза в сочетании с другими методиками (блот-гибридизации, авторадиографии). Дерматоглифика и пальмоскопия В 1892 г. Ф. Гальтоном в качестве одного из методов исследования человека был предложен метод изучения кожных гребешковых узоров пальцев и ладоней, а также сгибательных ладонных борозд. Он установил, что указанные узоры являются индивидуальной характеристикой человека и не изменяются в течение его жизни. Ф. Гальтон уточнил и дополнил классификацию рельефа кожных узоров, основы которой были разработаны Я. Пуркинье еще в 1823 г. Позднее классификацию Гальтона усовершенствовали ряд ученых; она и сейчас широко используется в криминалистике и генетических исследованиях. В настоящее время установлена наследственная обусловленность кожных узоров, хотя характер наследования окончательно не выяснен. Вероятно, этот признак наследуется по полигенному типу. На характер пальцевого и ладонного узоров организма большое влияние оказывает мать через механизм цитоплазматической наследственности. Дерматоглифические исследования важны при идентификации зиготности близнецов. Считают, что если из 10 пар гомологичных пальцев не менее 7 имеют сходные узоры, это указывает на однояйцевость. Сходство узоров лишь 4—5 пальцев свидетельствует в пользу разнояйцевости близнецов. Изучение людей с хромосомными болезнями выявило у них специфические изменения не только рисунков пальцев и ладоней, но и характера основных сгибательных борозд на коже ладоней. Характерные изменения этих показателей наблюдаются при болезни Дауна, при синдромах Клайнфельтера, Шерешевского — Тернера, что позволяет использовать методы дерматоглифики и пальмоскопии в диагностике этих заболеваний. Определяются специфические Дерматоглифические изменения и при некоторых хромосомных аберрациях, например при синдроме «кошачьего крика». Применяют эти методы и с целью установления отцовства. 99. Методы в генетике человека. Молекулярно-генетические методы (исследование ДНК). Генетическое тестирование. Генетическое прогнозирование. Локализация соответствующих повреждений в самом наследственном материале может быть выявлена методами молекулярной генетики. Разработка метода обратной транскрипции ДНК на молекулах иРНК определенных белков с последующим размножением этих ДНК привела к появлению ДНК-зондов для различных мутаций нуклеотидных последовательностей человека. Использование таких ДНК-зондов для гибридизации с ДНК клеток пациента дает возможность выявлять у него соответствующие изменения в наследственном материале, т.е. диагностировать определенные виды генных мутаций (генодиагностика). Важными достижениями молекулярной генетики последних десятилетий явились работы по секвенированию — определению нуклеотидной последовательности ДНК. В настоящее время полностью установлена последовательность нуклеотидов многих генов человеческого генома, в том числе генов α- и (β-глобиновых цепей гемоглобина, некоторых полипептидных гормонов (инсулина, гормона роста, хорионического соматотропина, пролактина). Интенсивно изучаются нуклеотидные последовательности генов актинов, тубулинов, интерферонов. Этими исследованиями выявлена высокая степень генетического полиморфизма у человека, который часто не проявляется фенотипически. Методы молекулярной генетики и генной инженерии позволяют не только диагностировать целый ряд генных мутаций и устанавливать нуклеотидную последовательность отдельных генов человека, но и размножать (клонировать) их и получать в большом количестве белки — продукты соответствующих генов. Клонирование отдельных фрагментов ДНК осуществляется путем включения их в бактериальные плазмиды, которые, автономно размножаясь в клетке, обеспечивают получение в большом количестве копий соответствующих фрагментов ДНК человека. Последующая экспрессия рекомбинантных ДНК в бактериях позволяет получить белковый продукт соответствующего клонированного человеческого гена. Таким образом, с помощью методов генной инженерии стало возможно получать на основе человеческих генов некоторые первичные генные продукты (инсулин). Это определяет перспективы терапии наследственных болезней, обусловленных связанным с генными мутациями дефицитом нормальных продуктов генов. Дальнейшее совершенствование методов молекулярной генетики обеспечит возможность полного определения нуклеотидных последовательностей не только структурных, но и регуляторных локусов генома человека, а разработка методов включения в человеческий геном нормальных нуклеотидных последовательностей в перспективе может стать основой генотерапии. Генетическое тестирование, генетический тест или ДНК-тест - это современная методика, использующая сложные технические средства для исследования именно молекулы ДНК. Генетический тест используется преимущественно для диагностики генетических заболеваний, однако применение может быть гораздо шире. Существуют методы, которые также используются для выявления продуктов генов – например, белков, ферментов и т.д. Отдельные методы позволяют изучать микроструктуру хромосом. Генетический тест может быть использован с целью: • скрининга носителей, в частности идентификации лиц, не имеющих выраженного генетического заболевания, но могут иметь одну копию гена, ответственного за возникновение этого генетического заболевания, тогда как для развития этого заболевания необходимо наличие двух копий; • предимплантационой генетической диагностики (скрининга эмбриона на наследственные заболевания); • пренатальной диагностики; • скрининга новорождённых; • досимптоматического исследования у лиц, развитие заболевания может произойти в старшем возрасте, например болезни Хантингтона; • пресимптоматического исследования риска возникновения в будущем полигенных заболеваний, рака или, например болезни Альцгеймера; • подтверждение диагноза генетического заболевания у симптоматического пациента; • для судебной экспертизы, напр. идентификации личности. 100. Генетическая гетерогенность популяций в человеческом обществе. Популяционно-статистический метод. С помощью популяционно-статистического метода изучают наследственные признаки в больших группах населения, в одном или нескольких поколениях. Существенным моментом при использовании этого метода является статистическая обработка получаемых данных. Этим методом можно рассчитать частоту встречаемости в популяции различных аллелей гена и разных генотипов по этим аллелям, выяснить распространение в ней различных наследственных признаков, в том числе заболеваний. Он позволяет изучать мутационный процесс, роль наследственности и среды в формировании фенотипического полиморфизма человека по нормальным признакам, а также в возникновении болезней, особенно с наследственной предрасположенностью. Этот метод используют и для выяснения значения генетических факторов в антропогенезе, в частности в расообразовании. При статистической обработке материала, получаемого при обследовании группы населения по интересующему исследователя признаку, основой для выяснения генетической структуры популяции является закон генетического равновесия Харди — Вайнберга. Он отражает закономерность, в соответствии с которой при определенных условиях соотношение аллелей генов и генотипов в генофонде популяции сохраняется неизменным в ряду поколений этой популяции. На основании этого закона, имея данные о частоте встречаемости в популяции рецессивного фенотипа, обладающего гомозиготным генотипом (аа), можно рассчитать частоту встречаемости указанного аллеля (а) в генофонде данного поколения. Распространив эти сведения на ближайшие поколения, можно предсказать частоту появления в них людей с рецессивным признаком, а также гетерозиготных носителей рецессивного аллеля. Математическим выражением закона Харди — Вайнберга служит формула (рА. + qa)2, где р и q — частоты встречаемости аллелей А и а соответствующего гена. Раскрытие этой формулы дает возможность рассчитать частоту встречаемости людей с разным генотипом и в первую очередь гетерозигот — носителей скрытого рецессивного аллеля: p2AA + 2pqAa + q2аа. Например, альбинизм обусловлен отсутствием фермента, участвующего в образовании пигмента меланина и является наследственным рецессивным признаком. Частота встречаемости в популяции альбиносов (аа) равна 1:20 000. Следовательно, q2 = 1/20 000, тогда q = 1/141, p+q=14 p=1 – 1/141; p = 140/141. В соответствии с формулой закона Харди — Вайнберга частота встречаемости гетерозигот = 2pq, т.е. 2pq(Аа)=2*140/141*1/141=1/70. Это означает, что в данной популяции гетерозиготные носители аллеля альбинизма встречаются с частотой один на 70 человек. |