генетика. генетика рк 2. 2 Аралы баылау сратары Онтогенез. Азаны жеке дамуы. Онтогенез кезедері
 Скачать 0.99 Mb. Скачать 0.99 Mb.
|

|
Эдвардс синдромы. Кариотип - 47, XX, 18 Тарату - 1: 5000 - 1: 7000 Бұл ауруды 1960 жылы Дж.Эдвардс айқындап тапқап.Бұл аурумен ерлерге қарағанда әйелдер көбірек ауырады. Аурдың негізі сипаттамалары:нәрестелердің салмағы өте жеңіл,бойлары кішкентай болуы,иектері тегіс,жақтары нашар дамыған ,бас сүйегі,құлақтары кішкентай және бас сүйегіне төмендеу орналасқан,тұмсықтары шығыңқы құстұмсық болып келеді.Көздерінің мөлдір қабығының бұдырлануы,көру нерв дискісінің семуі сияқты көру мүшелерінің мүкістігі байқалады.Қол саусақтары өте ұзын немесе қысқа болып,2-5 саусақтары ерекше орналасқан.Табандарының пішіні өзгеше.Эдвардс синдромын нәресте туылған кезде,плацентаның кішкентай болуы және жалғыз кіндік артериясының болуыарқылы күні бұрын анықтауға болады. Мысық айғайы синдромы. Кариотип - 46, ХХ 5р- Таралуы – жиілігі- 1:45000. Этиологиясы, патогенезі - цитогенети- калық варианттары 5 хромосоманың қысқа иығының делециясымен байланысты екені дәлелденген. Синдромның негізгі белгілері дамуы үшін - 5р15 сегментінің маңызы зор. Қарапайым делециядан басқа 5 сақиналы хромосомалар, мозаикалық формалары, сондай- ақ, 5- хромосоманың қысқа иығының арасындағы транслокация (критикалық сегментті жоғалтуы) және басқа аутосомалар байқалған. Фенотиптік белгілері балалардың жылағандағы даусы мысықтың мияулағанына немесе айқайлағанына ұқсайды. Бұл көмейдің немесе дыбыс байламдарының патологиясымен байланысты. «Мысықша мияулау» синдромы бар балалардың ақыл-естерінің дамуы үдемелі болғанымен, өздерінің жасындағылардан қатты қалыңқы болады. Туған кезде дене салмағының төмен болуы. Ақыл-ес дамуының тежелуі, Жұтыну қиындығыЖылаған кезде даусының мысықтың мияулағанына ұқсауы, ларингомаляция, микроцефалия, қылилық, Құлақтарының кескінсіздігі. Жүректің және басқа да ішкі ағзалардың туа біткен даму ақаулары, сүйек-бұлшықет жүйесінің өзгерістері, бұлшықеттік гипотония кездеседі. Науқастардың өмір суру ұзақтығы ішкі ағзалардың туа біткен ақауларына байланысты. Науқас балалардың көпшілігі бірінші жылы өлсе шамамен 10%-ы 10 жасқа дейін өмір суреді. Шерешевский-Тернер синдромы. Кариотип- (45, Х0) Таралуы - жиілігі 1:2000-1:5000. Этиологиясы, патогенезі – эмбрион-да біріншілік жыныс жасушаларының саны қалыпты бастама алады, бірақ жүктіліктің екінші жартысында олардың тез инволюциясы (кері даму) өтіп, бала туылар кезде жұмыртқадағы фоллику-лалар саны қалыптымен салыстырғанда күрт азаяды немесе мүлдем болмай-ды. Бұл әйел жыныс гормондарының айқын жеткіліксіздігіне, жыныстық жетілмеуге, науқастардың көпшілігінде – біріншілік аменореяға (менструация-ның болмауы) және бедеулікке әкеледі. Пайда болған хромосомалық бұзылыстар даму ақауларының себебі болып табылады. Фенотиптік белгілері – синдром үш бағытта көрінеді: 1) гипогонадизм, жыныс ағзаларының жеткіліксіздігі және екіншілік жыныс белгілері; 2) туа біткен даму ақаулары; 3) бойының аласа болуы. Жыныс жүйесі жағынан гонаданың болмауы, жатыр мен жатыр түтіктерінің гипоплазиясы, біріншілік аменорея, қасаға мен қолтық түгінің аз болуы, сүт бездерінің дамымауы байқалады. Мой-ындары қысқа, тері қатпарлары көп, қол-аяқ бастарының лимфалық ісінуі, бойының өспеуі (135 см) байқалады, интеллекті сақталады. Бойы ала-са, жүрек ақаулықтары, бүйректер дамуының ақаулары, маңдайының биік болуы байқалып, мойнында қанат тәрізді қатпарлар дамыған болады. Клайнфельтер синдромы. Кариотип – 47, XX Таралуы - 1:500 – 1:700. Этиологиясы, патогенезі – ер балаларда нәтижесінде жыныстық жетілу бұзылатын артық X хромосома (минимум біреу) болуымен сипатталатын патология. Синдромның негізгі сипаты:бойлары өте ұзын,иықтары тар,бөкселері кең,бұлшықеттері нашар дамыған,астеник немесе әтек типтес болып келеді.Беттерінде және қолтықтарында мардымсыз,өте сирек түктері болады.Бұл синдроммен ауырған адамдардың дерматоглификациясы өзгерген-қол саусақтарының өрнегінде доғалар жиі кездесіп,қырлар саны азаяды. 13. Адамда жыныс хромосомалар санының бұзылуынан болатын хромосомалық аурулар. Шерешевский-Тернер синдромы. Кариотип- (45, Х0) Таралуы - жиілігі 1:2000-1:5000. Этиологиясы, патогенезі – эмбрион-да біріншілік жыныс жасушаларының саны қалыпты бастама алады, бірақ жүктіліктің екінші жартысында олардың тез инволюциясы (кері даму) өтіп, бала туылар кезде жұмыртқадағы фолликулалар саны қалыптымен салыстырғанда күрт азаяды немесе мүлдем болмайды. Бұл әйел жыныс гормондарының айқын жеткіліксіздігіне, жыныстық жетілмеуге, науқастардың көпшілігінде – біріншілік аменореяға (менструация-ның болмауы) және бедеулікке әкеледі. Пайда болған хромосомалық бұзылыстар даму ақауларының себебі болып табылады. Фенотиптік белгілері – синдром үш бағытта көрінеді: 1) гипогонадизм, жыныс ағзаларының жеткіліксіздігі және екіншілік жыныс белгілері; 2) туа біткен даму ақаулары; 3) бойының аласа болуы. Жыныс жүйесі жағынан гонаданың болмауы, жатыр мен жатыр түтіктерінің гипоплазиясы, біріншілік аменорея, қасаға мен қолтық түгінің аз болуы, сүт бездерінің дамымауы байқалады. Мой-ындары қысқа, тері қатпарлары көп, қол-аяқ бастарының лимфалық ісінуі, бойының өспеуі (135 см) байқалады, интеллекті сақталады. Бойы ала-са, жүрек ақаулықтары, бүйректер дамуының ақаулары, маңдайының биік болуы байқалып, мойнында қанат тәрізді қатпарлар дамыған болады. Клайнфельтер синдромы. Кариотип – 47, XX Таралуы - 1:500 – 1:700. Этиологиясы, патогенезі – ер балаларда нәтижесінде жыныстық жетілу бұзылатын артық X хромосома (минимум біреу) болуымен сипатталатын патология. Мозаикалық формалары - 46ХУ/47ХХУ айтарлықтай сирек кездеседі. Көп жағдайда (шамамен 67%) Клайнфельтер синдромы бірінші немесе екінші мейоздық бөліну кезінде Ххромосоманың ажырамауынан немесе зигота дамуы кезеңінде хромосоманың митоздық ажырауының бұзылысы себебінен бо лады (мозаикалық варианттары). Фенотиптік белгілері – бойларының ұзын болуы, дене бітімінің астениялық нәзіктігі, аталық бездер гипоплазиясы, импотенция және бедеулік, сүт бездерінің ісінуі, жамбастарының жалпақ болуы, алақанда көлденең қатпар болуы, ересектерде семіздік және алкоголизмге бейімділік болады, ақыл-ес дамуы аздап төмендеген, жыныс ағзалары кішкентай, гинекомастия, тестостерон деңгейінің төмен болуы, гонадотропин деңгейінің жоғары болуы, бетте түк өсуінің нашарлауы. Науқастар бедеу болады. 14.Мультифакторлық аурулар. Тұқым қуалау ерекшелігі. Генокопия және фенокопия. Мультифакторлы аурулар – бұл тұқым қуалайтын факторлардың (моно- немесе полигенді) орта факторлармен әсерінің нәтижесі. Бұл аурулардың ауру көріністері мен ағымында жас пен жынысқа байланысты айырмашылықтар болады. Адамның фенотиптік белгілерінің көпшілігі гендердің көптеген санымен бақыланады. Бұл гендердің әрқайсысы басқаларына байланыссыз әсер етеді. Гендердің қалыпты таралуында сыртқы орта факторларының нақты әсері бар. Көптеген жағдайларда популяцияда фенотиптік белгілердің өзгергіштігі гендер мен сыртқы орта факторлары жиынтығының біріккен әсерін көрсетеді. Мультифакторлық генетикалық ауруларда барлық уақытта бір-бірімен кумуляциялық әсерлесетін гендер реттілігінен тұратын полигенді компонент болады. Адамның көптеген кең таралған аурулары: атеросклероз, жүректің ишемиялық ауруы, эссенциальды гипертония, бронхиальды демікпе, қант диабеті, псориаз, шизофрения, эпилепсия, дамудың оқшауланған ақаулары менделдік типпен тұқым қуаламайды, алайда, жанұяда аурудың жинақталуы, ауру ер кісілер мен әйелдер арасындағы айырмашылық, моно- және дизиготты егіздерде конкорданттылық, олардың дамуында тұқымқуалаушылықтың белгілі бір ролін көрсетеді. Бұл аурулар көптеген гендердің қатысуынан пайда болады, сондықтан оларды полигенді аурулар (мультифакторлы)деп атайды. Полигенді (мультифакторлық) аурулар тек генетикалық және сыртқы (қолайсыз) факторлардың бірлескен әсері жағдайында ғана пайда болады. Мультифакторлық аурулар генетикалық полиформизм феноменіменсипатталады. Олардың бірі бір (басты) геннің және басқа гендердің қосымша әсері салдарынан дамиды; басқалары бірнеше гендердің аддитивті (қосындылық) әсерінен дамиды. Генетикалық полиморфизм. Полиморфты деп популяцияда бірнеше түрмен – аллелдермен келтірілген гендер аталады. Бір геннің бір аллелдерінің арасындағы айырмашылық әдетте, оның генетикалық кодының аздаған вариациясында болады, сондай-ақ, соңғылары фенотиптік деңгейде көрінбеуі де, көрінуі де (клиникалық көріністерге дейін) мүмкін. Нақты аллелдердің қолайсыз дамуында әртүрлі аурулардың даму қатері жоғарылайды. Нуклеотидті реттілік полиморфизмі геномның барлық құрылымдық элементтерінде: экзондарда, интрондарда, реттеуші аймақтарда және т.б. анықталған. Полиморфизмнің көпшілік жағдайы не бір нуклеотид алмасуымен, не қайталанатын фрагменттер санының вариациясымен көрінеді. Тұқым қуалау бейімділігімен жүретін аурулардың полигенді табиғаты генеологиялық, егіздік және популяциялық-статистикалық әдістер көмегімен нақтыланады. Генетикалық бейімділік генотипте гендердің белгілі мөлшері жинақталған (полигендер) жағдайда пайда болып, аурудың даму шегін түзеді, бұл жағдайда ауру тек қолайсыз сыртқы орта факторлары әсер еткен жағдайда ғана фенотиптік (клиникалық) көрініс береді. Мультифакторлық аурулардың дамуының мұндай механизмі шектік әсермен мультифакторлық тұқым қуалау моделіне негіздеген. Тұқым қуалаудың мұндай моделіне сәйкес келуі гендердің (генотиптердің) ұқсастығына байланысты жанұяда науқастардың көп болуымен, науқастардың туысқандарының арасында оның жиілігінде айырмашылығының (I дәрежелі туыстар арасында жиілігі жоғары болуы, ал II және III дәрежелі туыстар арасында ауру жиілігінің төмендеуі) болуымен сипатталады. Туыстар арасында (жалпы популяциямен салыстырғанда) ауруға бейімдеуші гендердің көп мөлшерде жинақталуына байланысты аурудың ерте басталуы және аурудың клиникалық көрінісінің ауыр болуына тәуелді аурудың жиілігі артады. Мультифакторлық аурулар әйелдер мен ер кісілердің зақымдалу жиілігі әртүрлі болумен сипатталады, дәлірек айтқанда, аз зақымдалатын жыныс туыстарында жиі кездеседі, ал көп зақымдалатын жыныс туыстарында аз кездеседі. Бұл құбылыс аз зақымдалатын жыныста гендердің көп жиналуымен, және сәйкес, оның туыстарында гендердің көп жиналуымен түсіндіріледі. 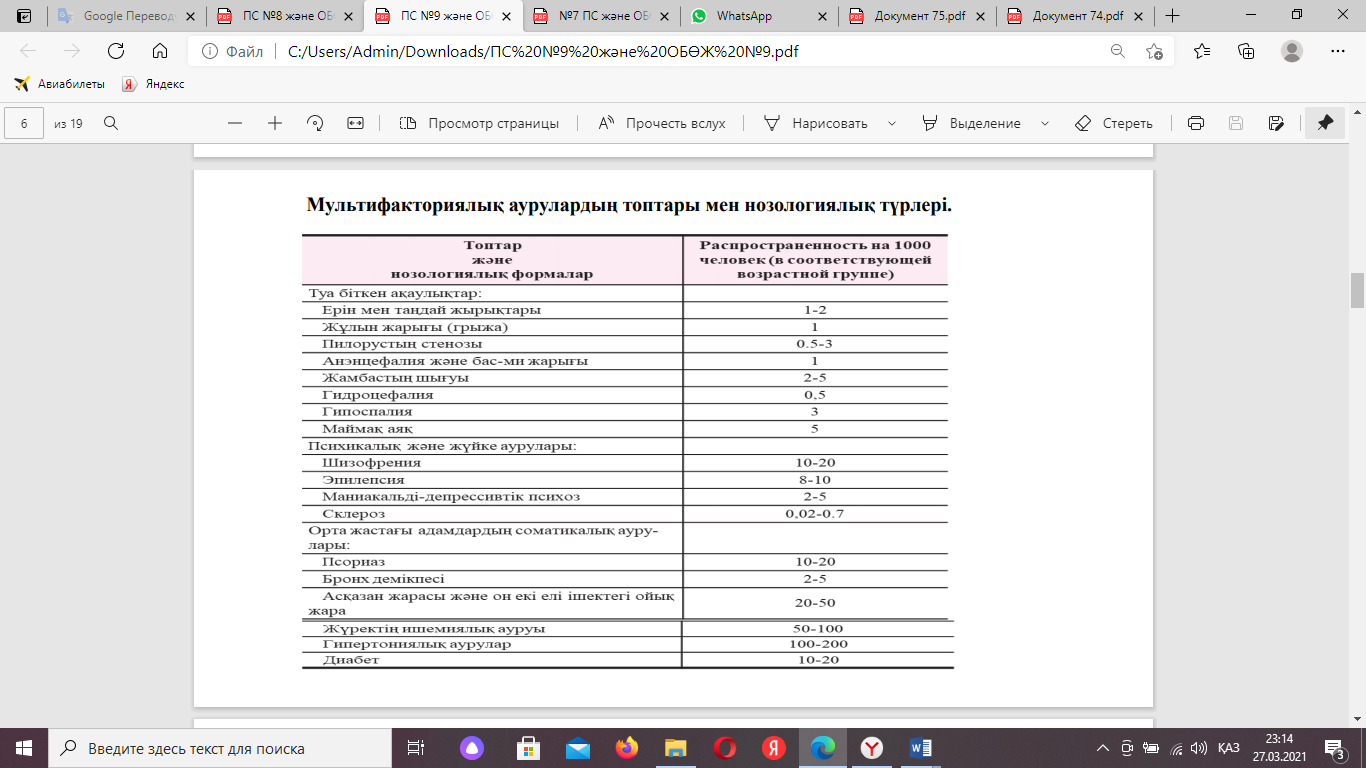 Барлық бейімдеуші факторлар: генетикалық және сыртқы орта факторлары қауіп факторлары деп жалпы түсінікке біріктіріледі. Бронхтық демікпе Орналасуы – 4, 5, 6, 7, 11, 13, 14 хромосомаларда 30-дан артық ген болады. Полигенді берілетін аурулардың классикалық моделі бронхтық демікпе болып табылады. Таралуы – әлемде 0,2-18%. Бронхтық демікпемен ауратын балалардың 50-86%-да аллергиялық ауруларға жанұялық қолайсыздық анықталады. Монозиготалы егіздерде бронхтық демікпенің даму қатері дизиготалы егіздерге қарағанда анағұрлым жоғары болады. Этиологиясы, патогенезі – а) генетикалық факторлар (бейімдеуші гендер); б) сыртқы факторлар (аллергендер, инфекция, шылым шегу, тамақ және т.б.). бронхтық демікпе негізінде иммундық механизмдер жататы- ны белгілі. Бронхтық демікпемен ауыратын науқастардың көпшілігінде әртүрлі аллер- гендерге арнамалы антидене анықталады. Бұл ауру негізінде бронхтардың жедел спазмы дамып, шырыш бөлудің күшеюіне әкелетін тыныс жолдарының аллергиялық қабынуы жатады. Аталған өзгерістер бронх- тар өткізгіштігінің бұзылысы - бронхтық обструкция тудырады. Фенотиптік белгілері – бронхтық демікпенің белгілері: жөтел – жиі, тұрақты, асқынған болуы мүмкін; Экспираторлы (тыныс шығарудың қиындауы басым болады) ентігу – сырылдың болуы; тұншығу. Қант диабеті Орналасуы – 6-шы хромосомада орналасқан бірқатар гендердің ақауы. Таралуы – жер шары тұрғындарының 4 %-ы. Этиологиясы, патогенезі – инсулин өндірілуіне жауап беретін ген мутациясы, ұйқы безінің эндокринді жасушаларының жалпы өлімі вирусты инфекцияларда, онкологиялық ауруларда, панкреатитте, ұйқы безінің уытты зақымдануларында, күйзелістік жағдайларда иммунды жүйе жасушалары ұйқы безінің β-жасушаларына қарсы антидене түзіп, оларды бұзатын әртүрлі аутоиммунды ауруларда кездеседі. Қант диабеті — негізгі диагностикалық белгісі созылмалы гипергликемия – қандағы қант деңгейінің жоғарылауы, полиурия, организмнің жалпы әлсіреуі болып табылатын эндокринді жүйенің аутоиммунды ауруы; іштегі ауру сезімі; ауру көріністері ұзақ уақыт болғанда және ауру диагностикасы болмағанда организмнің майлар ыдырауы өнімдерімен улануы басталады – жиі теріден, ауыздан ацетон иісі шығуымен белгілер береді. Фенотиптік белгілері – диабеттің І типінің симптомдары: қатты шөлдеу; адам тәулігіне 3-5 литрге дейін сұйықтық ішеді; тыныс шығарғанда ацетон исінің болуы; науқастың тәбеті жоғары болады, тамақты көп жейді, бірақ тез арықтайды; әсіресе түнде жиі және көп мөлшерде несеп шығару (полиурия деп аталады); жарақаттардың нашар жазылуы; терісінің қышуы, зең аурулары немесе фурункулалар жиі болады. Моногенді аурулармен салыстырғанда мультифакторлы ауруларға суммарлы әсерлі гендер санының айтарлықтай көп болуы мен сыртқы орта факторына байланысты болатын тұқым қуалау бейімділігі тән. Жүректің ишемиялық ауруы (ЖИА) – бұл жүрекке қан мен оттегіні жеткізетін шағын қан тамырларының (коронар тамырлары) тарылуынан пайда болатын ауру. Себептері: Жүректің ишемиялық ауруы ерлер мен әйелдер арасындағы өлімнің жетекші себебі болып табылады. Жүректің ишемиялық ауруының себебі болып коронар тамырланың атеросклерозы табылады. Бұл процесс коронарлы тамырларының қабырғаларының қатаюына алып келеді. Процесстің басында майлардан тұратын түйіншектер пайда болады. Олардың кесірінен қан тамырлары тарыла бастайды. Тарылған коронарлы тамырлар қажетті деңгейде жүрекке қанды жеткізе алмайды. Жүрек-қан тамырлар ауруларының қауіп қатер факторлары деп осы аурулардың даму қауіпін жоғарлататын факторларды атаймыз. Олар модификацияға бағынатын және бағынбайтын факторларға бөлінеді. Симптомдары: ауырсыну - аурудың пайда болу орыны, таралымы стенокардияға ұксас болып, көбіне төс немесе жүрек аймағында пайда болады. Төс немесе жүрек аймағы басып, сыздап, ашып аурады. Ауру әдетте сол қол, иық, мойын, жауырын кейде екі қолға да, жақ сүйегіне таралады. Демікпе; Шаршағандық; Жалпы әлсірегендік. Диагностикасы: Коронароангиография – бұл жүректің қан тамырларының рентген көмегімен инвазивті зерттеу тәсілі; Стресс – эхокардиография Электрокардиография (ЭКГ) Электронды-сәулелі компьютерлік томография (ЭСКТ) – кальцийдің артерия қабырғаларында жиналуын анықтайтын диагностика тәсілі; Жүректің компьютерлік томографиясы; Нуклеарлы стресс-тест Жүректің ишемиялық ауруының (ЖИА) пайда болуына қауіп факторлары ЖИА немесе басқа жүрек қан тамырлар ауруларымен туыстарының ауруы, төменгі тығыздықты холестериннің мөлшерінің жоғарылауы АpoB жоғары деңгейі липопротеин, фибриногеннің қан сарысуында жоғарылауы, гипертония, семіру, гомоцистеиннің мөлшерінің көбеюі, майлы тағамды пайдалану, гиподинамия, темекі шегу және т.б.жатады. ЖИА сирек кездесетін түрі – отбасылық ақауы Аро-В-100. Аро-В геніндегі мутациялар, 2р23-р24. Альцгеймер ауруы –ересек адамдарда жиі кездесетін нейродегенеративтік ауру. Ол көбінесе деменцияның дамуына (есте сақтаудың жойылуы) алып келеді. деменцияның ең кең таралған түрі 1906 жылы неміс психиатры Алоис Альцгеймер тарапынан сипатталған нейрогенеративті сырқат. Әдетте 65 жастан асқан адамдарда кездеседі. Жасы ұлғая келе кейбір адамдар ұмытшақ бола бастайды. Қартая келе тіпті бала сияқты болып кететіндер де кездеседі. Қариялардағы бұл құбылысты біз «алжыды» деп жатамыз. Ал медицинада ол Альцгеймер ауруы деп аталады. Клиникалық симптомдары : есте сақтау қабілетінің күрт төмендеуі, сөйлеу, жазу, кеңістікте бағдарлау қабілеттерінің бұзылуы, үделеп дамушы жарыместік, эпилептикалық ұстамалар. Аурудың екі формасы бар: ерте басталатын (40-58 жас), кеш басталатын (58-80 жас). Аурудың үш моногендік нұсқаларының дамуына алып келетін 3 ген мутациялары да анықталды: Олардың біреуі АРР 21 хромосомада 21q 21 аймағында орналасқан және ол ми заттарында амилоидтық өсінділердің қалыптасуына қатыгнасатын B-амилоид ақуыздарының бастамасын кодтайды. Қалған екі ген – 14 хромосомада 14 q24, 3 және 1 хромосоманың 1q 31-42 локустарында орналасқан, тек нейрондарда экспрессияланатын және кейбір мембраналық ақуыздар – пресенилиндерді кодтайды. Диагностика Неврологиялық тексеру МРТ Интеллектуалды қабілетті тестілеу Қан талдауы |