Биохимия. ЭКЗАМЕНАЦИОННЫЕ ВОПРОСЫ ПО БИОЛОГИЧЕСКОЙ ХИМИИ. Экзаменационные вопросы по биологической химии для студентов лечебного, педиатрического и медикопрофилактического факультетов
 Скачать 6.22 Mb. Скачать 6.22 Mb.
|

|
Недостаточность фруктокиназыклинически не проявляется. Фруктоза накапливается в крови и выделяется с мочой, где её можно обнаружить лабораторными методами. Очень важно не перепутать эту безвредную аномалию с сахарным диабетом. Данное заболевание известно как доброкачественная эссенциальная фруктозурия и встречается с частотой 1:130 000. Наследственная непереносимость фруктозы,возникающая при генетически обусловленном дефекте фруктозо-1-фосфатальдолазы, не проявляется, пока ребёнок питается грудным молоком, т.е. пока пища не содержит фруктозы. Симптомы возникают, когда в рацион добавляют фрукты, соки, сахарозу. Рвота, боли в животе, диарея, гипогликемия и даже кома и судороги возникают через 30 мин после приёма пищи, содержащей фруктозу. У маленьких детей и подростков, продолжающих принимать фруктозу, развиваются хронические нарушения функций печени и почек. Непереносимость фруктозы - достаточно частая аутосомно-рецессивная форма патологии. Дефект альдолазы фруктозе-1-фосфата сопровождается накоплением фруктозе-1-фосфата, который ингибирует активность фосфоглюко-мутазы, превращающей глюкозо-1-фосфат в глюкозо-6-фосфат и обеспечивающей включение продукта гликогенфосфорилазной реакции в метаболизм. Поэтому происходит торможение распада гликогена на стадии образования глюкозо-1 -фосфата, в результате чего развивается гипогликемия. Как следствие, ускоряется мобилизация липидов и окисление жирных кислот. Следствием ускорения окисления жирных кислот и синтеза кетоновых тел, замещающих энергетическую функцию глюкозы, может быть метаболический ацидоз, так как кетоновые тела являются кислотами и при высоких концентрациях снижают рН крови. Результатом торможения гликогенолиза и гликолиза является снижение синтеза АТФ. Кроме того, накопление фосфорилированной фруктозы ведёт к нарушению обмена неорганического фосфата и гипофосфатемии. Для пополнения внутриклеточного фосфата ускоряется распад адениловых нуклеотидов. Продукты распада этих нуклеотидов включаются в катаболизм, проходя стадии образования гипоксантина, ксантина и, наконец, мочевой кислоты. Повышение количества мочевой кислоты и снижение экскреции уратов в условиях метаболического ацидоза проявляются в виде гиперурикемии. Следствием гиперурикемии может быть подагра даже в молодом возрасте Глицеральдегид -3 –фосфат 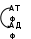  АТФ АДФ Нарушения метаболизма галактозы Обмен галактозы особенно интересен в связи с наследственным заболеванием - галактоземией. Галактоземиявозникает при нарушении обмена галактозы, обусловленном наследственным дефектом любого из трёх ферментов, включающих галактозу в метаболизм глюкозы
Галактоземия, вызванная недостаточностью галактозо-1-фосфатуридилтрансферазы (ГАЛТ), наиболее хорошо изучена. Это заболевание проявляется очень рано, и особенно опасно для детей, так как основным источником углеводов для них служит материнское молоко, содержащее лактозу. Ранние симптомы дефекта ГАЛТ: рвота, диарея, дегидратация, уменьшение массы тела, желтуха. Они появляются вскоре после рождения, как только ребёнок начинает получать молоко. В крови, моче и тканях повышается концентрация галактозы и галактозо-1-фосфата. В тканях глаза (в хрусталике) галактоза восстанавливается альдоредуктазой с образованием галактитола (дульцита). В этой реакции в качестве донора водорода используется NADPH. Восстановление галактозы происходит и в ходе нормального метаболизма, но протекает с небольшой скоростью. При галактоземии галактитол накапливается в стекловидном теле и связывает большое количество воды. Вследствие этого нарушается баланс электролитов, а чрезмерная гидратация хрусталика приводит к развитию катаракты, которая наблюдается уже через несколько дней после рождения. Тяжёлые последствия дефекта ГАЛТ наблюдают в печени. Это связано с накоплением галактозо-1-фосфата и его токсическим действием на гепатоциты. В результате возникают нарушения функции печени: гепатомегалия, жировая дистрофия. В почках таких больных также повышена концентрация галактитола и галактозо-1-фосфата, что влияет на их функции. Отмечают нарушения в клетках полушарий головного мозга и мозжечка, в тяжёлых случаях - отёк мозга, задержку умственного развития, возможен летальный исход. Для галактоземии, вызванной дефектом галактокиназы, тоже характерна катаракта, но при этом заболевании, в отличие от дефекта ГАЛТ, не отмечают нарушений функций печени, почек, мозга. Наиболее тяжёлые последствия снижения активности ГАЛТ связывают с влиянием галактозо-1-фосфата на активность других ферментов, участвующих в углеводном обмене (фосфоглюкомутазы, глюкозо-6-фосфатдегидрогеназы). Известно несколько форм галактоземии, причиной которой является недостаточность ГАЛТ Некоторые дефекты в строении ГАЛТ приводят лишь к частичной потере активности фермента. Поскольку в норме ГАЛТ присутствует в организме в избытке, то снижение его активности до 50%, а иногда и ниже может клинически не проявляться. При диагностике галактоземии исследуют мочу на содержание галактозы, собранную после нескольких кормлений молоком. При обнаружении у ребёнка катаракты его обследуют на недостаточность галактокиназы и ГАЛТ. Наличие галактозы в моче при отсутствии нарушений функции печени указывает на дефект галактокиназы. При обследовании проведение теста с нагрузкой галактозой не рекомендуется, так как этот тест опасен для больных. Лечение заключается в удалении галактозы из рациона. 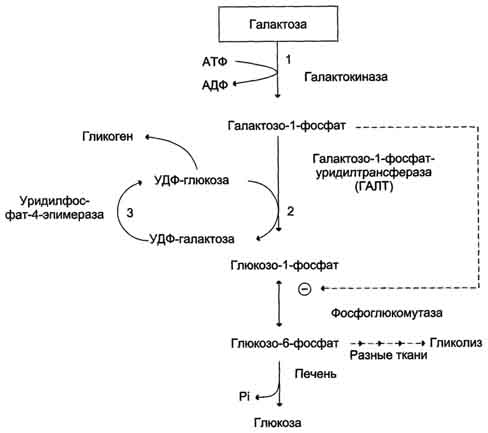 Классификация, биохимическая и генетическая характеристика гликогенов.
58. Важнейшие липиды тканей человека. Резервные липиды (жиры) и липиды мембран (сложные липиды). Жирные кислоты липидов тканей человека. Жирные кислоты- структурные компоненты различных липидов. В составе триацилгли-церолов жирные кислоты выполняют функцию депонирования энергии, так как их радикалы содержат богатые энергией СН2-группы. При окислении СН-связей энергии выделяется больше, чем при окислении углеводов, в которых атомы углерода уже частично окислены (-НСОН-). В составе фосфолипидов и сфинго-липидов жирные кислоты образуют внутренний гидрофобный слой мембран, определяя его свойства. Жиры и фосфолипиды организма при нормальной температуре тела имеют жидкую консистенцию, так как количество ненасыщенных жирных кислот преобладает над насыщенными. В фосфолипидах мембран ненасыщенных кислот может быть до 80-85%, а в составе жиров подкожного жира - до 60%. Жирные кислоты липидов человека представляют собой углеводородную неразветвлённую цепь, на одном конце которой находится карбоксильная группа, а на другом - метальная группа (ω-углеродный атом). Большинство жирных кислот в организме содержат чётное число атомов углерода - от 16 до 20 Состав жирных кислот подкожного жира человека
Жирные кислоты, не содержащие двойных связей, называют насыщенными. Основной насыщенной жирной кислотой в липидах человека является пальмитиновая (до 30-35%). Жирные кислоты, содержащие двойные связи, называют ненасыщенными. Ненасыщенные жирные кислоты представлены моноеновыми (с одной двойной связью) и полиеновыми (с двумя и большим числом двойных связей). Если в составе жирной кислоты содержатся две и более двойных связей, то они располагаются через СН2-группу. По положению первой двойной связи от метального углерода полиеновые жирные кислотыделят на семейства ω-3 и ω-6. Ацилглицеролы- сложные эфиры трёхатомного спирта глицерола и жирных кислот. Глицерол может быть связан с одной, двумя или тремя жирными кислотами, соответственно образуя моно-, ди- или триацилглицеролы (МАГ, ДАГ, ТАГ). Основную массу лигщдов в организме человека составляют триацилглицеролы - жиры. У человека с массой тела 70 кг в норме содержится до 10 кг жиров. Они запасаются в жировых клетках - адипоцитах и используются при голодании как источники энергии. Моно- и диацилглицеролы образуются на промежуточных этапах распада и синтеза триацил-глицеролов. Атомы углерода в глицероле по-разному ориентированы в пространстве, поэтому ферменты различают их и специфически присоединяют жирные кислоты у первого, второго и третьего атомов углерода. Фосфолипиды - разнообразная группа липидов, содержащих в своём составе остаток фосфорной кислоты. Фосфолипиды делят на глицерофосфолипиды, основу которых составляет трёхатомный спирт глицерол, и сфинго-фосфолипиды - производные аминоспирта сфингозина. Фосфолипиды имеют амфифильные свойства, так как содержат алифатические радикалы жирных кислот и различные полярные группы. Благодаря своим свойствам фосфолипиды не только являются основой всех клеточных мембран, но и выполняют другие функции: образуют поверхностный гидрофильный слой липопротеинов крови, выстилают поверхность альвеол, предотвращая слипание стенок во время выдоха. Некоторые фосфолипиды участвуют в передаче гормонального сигнала в клетки. Сфингомиелины являются фосфолипидами, формирующими структуру миелиновых оболочек и других мембранных структур нервных клеток. Глицерофосфолипиды. Структурная основа глицерофосфолипидов - глицерол. Глицерофосфолипиды (ранее используемые названия - фосфоглицериды или фосфоацилглицеролы) представляют собой молекулы, в которых две жирные кислоты связаны сложноэфирной связью с глицеролом в первой и второй позициях; в третьей позиции находится остаток фосфорной кислоты, к которому, в свою очередь, могут быть присоединены различные заместители, чаще всего аминоспирты. Если в третьем положении имеется только фосфорная кислота, то глицерофосфолипид называется фосфатидной кислотой. Её остаток называют "фосфатидил"; он входит в название остальных глицерофосфолипидов, после которого указывают название заместителя атома водорода в фосфорной кислоте, например фосфатидилэтаноламин, фосфатидилхолин и т.д Плазмалогены. Плазмалогены - фосфолипиды, у которых в первом положении глицерола находится не жирная кислота, а остаток спирта с длинной алифатической цепью, связанный простой эфирной связью.Характерный признак плазмалогенов - двойная связь между первым и вторым атомами углерода в алкильной группе. Плазмалогены бывают 3 видов: фосфатидальэтано-ламины, фосфатидальхолины и фосфатидаль-серины. Плазмалогены составляют до 10% фосфолипидов мембран нервной ткани; особенно много их в миелиновых оболочках нервных клеток. Сфинголипиды.Аминоспирт сфингозин, состоящий из 18 атомов углерода, содержит гидроксильные группы и аминогруппу. Сфингозин образует большую группу липидов, в которых жирная кислота связана с ним через аминогруппу. Продукт взаимодействия сфингозина и жирной кислоты называют "церамид". В церамидах жирные кислоты связаны необычной (амидной) связью, а гидроксильные группы способны взаимодействовать с другими радикалами. Церамиды отличаются радикалами жирных кислот, входящих в их состав. Обычно это жирные кислоты с большой длиной цепи - от 18 до 26 атомов углерода. |