Глик Молекулярная биотехнология. Глик Б., Пастернак Дж. Молекулярная биотехнология. Принципы и применение. Пер с англ. М. Мир, 2002. 589 с
 Скачать 9.74 Mb. Скачать 9.74 Mb.
|

Системы ДНК-диагностикиИнформация о всем многообразии свойств организма заключена в его генетическом материале. Так, патогенность бактерий определяется наличием у них специфического гена или набора генов, а наследственное генетическое заболевание возникает в результате повреждения определенного гена. Сегмент ДНК, детерминирующий данный биологический признак, имеет строго определенную нуклеотидную последовательность и может служить диагностическим маркером. В основе многих быстрых и надежных диагностических методов лежит гибридизация нуклеиновых кислот — спаривание двух комплементарных сегментов разных молекул ДНК. Процедура в общих чертах состоит в следующем. 1. Фиксация одноцепочечной ДНК-мишени на мембранном фильтре. 2. Нанесение меченой одноцепочечной ДНК-зонда, которая при определенных условиях (температуре и ионной силе) спаривается с ДНК-мишенью. 3. Промывание фильтра для удаления избытка несвязавшейся меченой ДНК-зонда. 4. Детекция гибридных молекул зонд/мишень. В диагностических тестах, основанных на гибридизации нуклеиновых кислот, ключевыми являются три компонента: ДНК-зонд, ДНК-мишень и метод детекции гибридизационного сигнала. Система детекции должна быть в высшей степени специфичной и высокочувствительной.
188 ГЛАВА 9 Гибридизационные зондыЧтобы обеспечить адекватность диагностического теста, гибридизационные ДНК- и РНК-зонды должны быть высокоспецифичными. Другими словами, необходимо, чтобы зонд гибридизовался только с искомой нуклеотидной последовательностью. Если есть вероятность получения ложноположительного (наличие гибридизационного сигнала в отсутствие последовательности-мишени) или ложноотрицательного (отсутствие сигнала при наличии последовательности-мишени) результата, то целесообразность применения теста значительно снижается. Специфичность зондов может проявляться на разных уровнях: они могут «различать» два и более вида, отдельные штаммы в пределах одного вида или разные гены. В зависимости от ситуации зонды могут быть представлены молекулами ДНК или РНК; они могут быть длинными (более 100 нуклеотидов) или короткими (менее 50 нуклеотидов), представлять собой продукт химического синтеза, клонированные интактные гены или их фрагменты. Зонды получают разными способами. Один из них состоит в следующем, ДНК патогенного микроорганизма расшепляют с помощью рест-ринируюшей эндонуклеазы и клонируют в плазмидном векторе. Затем проводят скрининг рекомбинантных плазмид с использованием геномной ДНК как патогенного, так и непатогенного штаммов. Те плазмиды, которые содержат последовательности, гибридизующиеся только с ДНК патогенного штамма, составляют основу вилоспепифичных зондов. После этого проводят ряд дополнительных гибридизаций с ДНК, выделенными из различных организмов, чтобы удостовериться, что потенциальные зонды не дают с ними перекрестной гибридизации. Для определения чувствительности метода каждый из зондов проверяют также на модельных образцах, в том числе и на смешанных культурах. Весьма желательно, чтобы ДНК-диагностику можно было проводить на исходном материале, без дополнительного его культивирования или выделения нуклеиновых кислот, особенно в тех случаях, когда тестируются клинические образцы. Исследователи с успехом проводят гибридизацию с ДНК-мишенями, присутствующими в образцах кала, мочи, крови, смывах из зева и в тканях без предварительной их очистки. Если концентрация последовательности-мишени в исследуемом образце слишком мала, ее можно амплифицировать с помощью полимеразной цепной реакции (ПЦР). Диагностика малярииВ качестве примера использования ДНК-зондов для диагностики заболеваний можно привести процедуру обнаружения Plasmodiumfalciparum. Этот паразит вызывает малярию, заболевание, угрожающее примерно трети всего населения Земли. Он инфицирует эритроциты и разрушает их, что приводит к развитию лихорадки, а в тяжелых случаях — к поражению мозга, почек и других органов. Чтобы выявить источники инфекции, оценить эффективность мер по их ликвидации и обеспечить раннюю диагностику и лечение, необходимы достаточно чувствительные, простые и недорогие методы. В настоящее время малярию диагностируют с помощью микроскопического исследования мазков крови -эффективного, но трудоемкого и занимающего много времени процесса. Иммунологические методы обнаружения Plasmodium, такие как ELISA, достаточно быстрые и их легко автоматизировать, но с их помощью нельзя отличить текущую инфекцию от прошедшей, поскольку при этом определяется только наличие антител к Plasmodium в крови больных. Для избирательной ДНК-диагностики текущей инфекции, т. е. для выявления ДНК возбудителя, в качестве основы используются высокоповторяющиеся последовательности ДНК P. falciparum. Сначала с помощью ДНК-зонда проводится скрининг библиотеки геномной ДНК паразита. Затем отбираются клоны, дающие наиболее интенсивный гибридизационный сигнал, поскольку именно они предположительно содержат высокоповторяющиеся последовательности. ДНК каждого из отобранных клонов проверяют на способность к гибридизации с ДНК видов Plasmodium, не вызывающих малярию. В качестве специфического зонда выбирается последовательность, гибридизующаяся с ДНК Р. falciparum, но не с ДНК P. vivax, P. cynomolgi или с ДНК человека. С его помощью Молекулярная диагностика 189
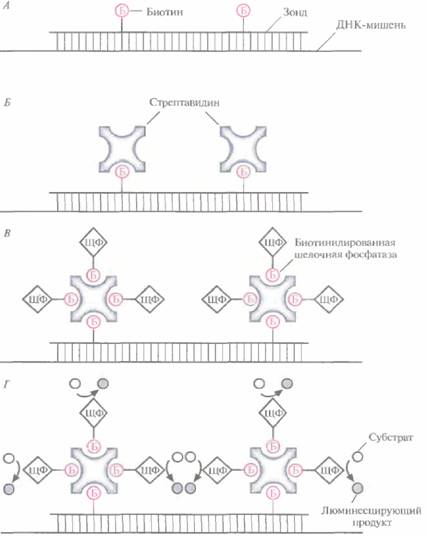
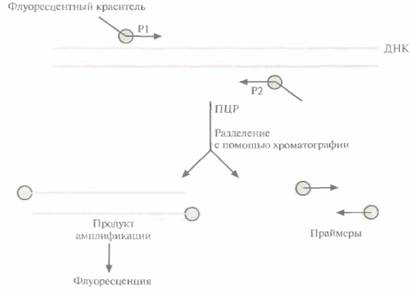
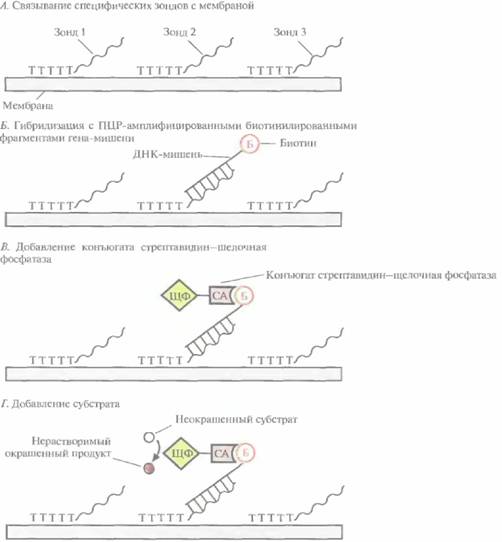
можно обнаруживать всего 10 пг очищенной ДНК P.falcipantm или 1 нг той же ДНК в крови больного. Получены и охарактеризованы более 100 различных ДНК-зондов, позволяющих обнаруживать патогенные штаммы различных бактерий, вирусов и паразитических простейших. Так, имеются зонды для диагностики бактериальных, инфекций человека, вызываемых Legionella рпеи-mophïla (респираторные заболевания). Salmonellatyphi{пищевые отравления), Campylobacterhyoin-testinaiis (гастриты), а также для выявления энте-ротоксичного штамма Escherich'ia colî (гастроэнтериты). Однако это лишь «верхушка айсберга»; в принципе с помощью гибридизации можно выявлять практически любые патогенные микроорганизмы. Выявление Trypanosoma cruziПаразитическое простейшее Tiypanosornacruziвызывает болезнь Чагаса (южноамериканский трипаносомоз), уносящую ежегодно примерно 50 тыс. жизней. Паразит широко распространен в Латинской Америке. Он переносится клопами-хищнецами, проникает в печень, селезенку, лимфатические узлы, центральную нервную систему и, размножаясь, разрушает клетки, в которых паразитирует. Для диагностики острой формы болезни Чагаса обычно проводят микроскопическое исследование свежей пробы периферической крови. Можно использовать и другой тест, более длительный, но выявляющий паразитов с большей вероятностью. Незараженных насекомых кормят кровью пациента и через 30—40 суг исследуют под микроскопом их кишечник на предмет наличия паразитов. Оба метода весьма трудоемки, дорогостоящи и требуют длительного времени. Болезнь можно диагностировать и иммунологическими методами, однако они часто дают ложноположительные результаты. В качестве альтернативы этим менее чем удовлетворительным процедурам было разработано несколько подходов, основанных на применении ПЦР. В насто- 190 ГЛАВА 9 ящее время ПЦР-диагностика болезни Чагаса служит дополнением к традиционным, широко используемым методам. Один из ПЦР-тестов основан на выявлении фрагмента ДНК длиной 188 п. н., который присутствует во множестве копий в геноме Т. cruzi, но отсутствует в геномной ДНК нескольких родственных паразитов. После амплификации этот фрагмент без труда обнаруживается с помощью электрофореза в полиакриламид-ном геле. Незначительно варьируя методику проведения ПЦР (например, изменяя нуклеотидную последовательность праймеров), последнюю можно использовать для обнаружения широкого спектра бактерий, вирусов и паразитов. Нерадиоактивные методы детекцииВ большинстве лабораторий для гибридизации используют зонды, меченные каким-либо радиоактивным изотопом, чаще всего 32Р. Такие зонды обладают высокой удельной радиоактивностью и обеспечивают хорошее отношение сигнал/шум. Радиоактивно меченный зонд наносят на фильтр с фиксированной на нем ДНК-мишенью, проводят гибридизацию, отмывают несвязавшуюся ДНК-зонд и детектируют метку с помощью радиоавтографии. Однако 32P является короткоживущим изотопом, испускающим высокоэнергетическое излучение; при работе с ним необходимо использовать специальное оборудование и обеспечивать безопасную утилизацию отходов. Чтобы обойти эти трудности, были созданы нерациоактивные системы детекции. Для усиления гибридизаци-онного сигнала в этом случае используется ферментативное превращение хромогенного или хемилюминесцентного субстрата: первый из них под действием фермента изменяет окраску, а второй испускает свет. В большинстве подобных систем применяются ДНК-зонды, содержащие биотинилированные нуклеотиды. Гибридизация и детекция сигнала проводятся более или менее стандартным образом. 1. Зонд, меченный биотином, гибридизуют с ДНК-мишенью (рис. 9.4, А). 2. Промывают фильтр для удаления избытка несвязавшегося зонда. 3. Добавляют авидин (белок куриного яйца) или стрептавидин (бактериальный аналог авидина) (рис. 9.4, Б). 4. Добавляют биотинилированный фермент щелочную фосфатазу или пероксидазу хрена (рис. 9.4, В). 5. В зависимости от используемого фермента добавляют хромогенный или хемилюминесцентный субстрат и регистрируют изменение окраски либо люминесценцию, сопровождающую превращение субстрата в продукт (рис. 9,4, Г). В качестве альтернативы после гибридизации ДНК с биотинилированным зондом можно добавлять уже готовый комплекс стрептавидин—фермент, имеющий сайт связывания с биотином. Как авидин, так и стрептавидин связываются с биотином очень прочно (константа диссоциации (Кd = 1015); кроме того, каждый из белков имеет четыре независимых биотинсвязывающих сайта, благодаря чему одна молекула авидина или стрептавидина может одновременно присоединять фермент и зонд, меченные биотином. Биотинилирование и связывание со стрептавидином не приводят к снижению ферментативной активности. В хромогенных системах детекции в том месте, где находится гибридная ДНК, под действием фермента образуется нерастворимый краситель, а в хемилюминесцентных системах — продукт, который испускает свет. Нерадиоактивные системы детекции обладают и другими преимуществами: биотинилированная ДНК остается стабильной при комнатной температуре как минимум год; методы регистрации хемилюминешенции обладают такой же чувствительностью, как и методы регистрации радиоактивного сигнала; детекция испускаемого света при помощи рентгеновской пленки или люминометра, как и регистрация изменения цвета, занимают несколько часов. По-видимому, хемилюминесцентные системы регистрации сигнала, все же более чувствительные, чем хромогенные, вскоре вытеснят все остальные, использующиеся при ДНК-диагностике. Если при этом применяется ГЩР, то амплифицируемый продукт можно пометить флуоресцентным красителем, присоединяя его Молекулярная диагностика 191
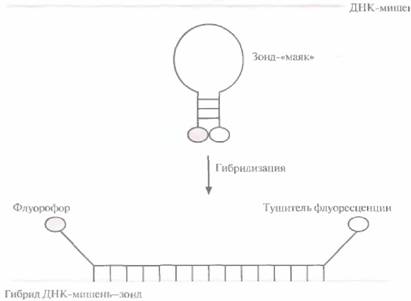
к 5'-концу каждого праймера. В качестве красителей часто используют флуоресцеин и родамин, которые испускают зеленый и красный свет соответственно. После ПЦР-амплификации ДНК-мишени проводят разделение флуоресцеин-ме-ченного праймера и продуктов амплификации, после чего регистрируют включение метки (рис. 9.5), Если ДНК-мишень в образце отсутствует, то не будет образовываться и флуоресцирующий продукт. Один из недавно разработанных нерадиоактивных методов детекции основан на использовании зонда — «молекулярного маяка» (рис. 9.6). Такой зонд состоит из 25 нуклеотидов. Средние 15 из них комплементарны ДНК-мишени и не спариваются друг с другом, а 5 концевых нуклеотидов взаимно комплементарны и образуют шпильку. К 5'-концу присоединен флуоресцентный хромофор (флуорофор), а к 3'-концу — нефлуоресцентный хромофор (тушитель), на который передается энергия возбуждения флуорофора. В растворе при комнатной температуре «маяк» имеет такую конфигурацию, при которой флуорофор и тушитель находятся в тесном контакте, и флуоресценция флуорофора тушится. Когда же 15 средних нуклеотидов зонда гибридизуются с комплементарной последовательностью ДНК- или РНК-мишени, происходит пространственное разделение 192 ГЛАВА 9
флуорофора и тушителя, и зонд испускает свет. Температура реакционной смеси должна быть близка к комнатной, поскольку при ее повышении шпилька денатурирует, флуорофор и тушитель расходятся и происходит флуоресценция. Необходимо также, чтобы все 15 нуклеотидов зонда были комплементарны соответствующей последовательности ДНК- или РНК-мишени. Геномная дактилоскопияМетод геномной дактилоскопии (ДНК-типирование) часто используется в судебной медицине для идентификации биологических образцов. С его помощью можно доказать, что подозреваемый действительно совершил преступление, или, напротив, что он невиновен. Для проведе-
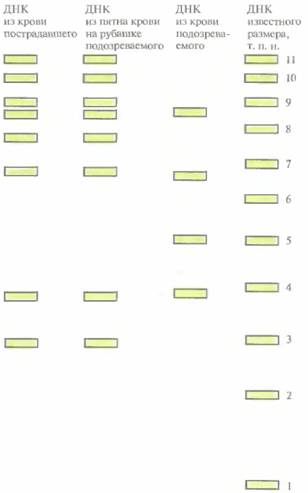
Молекулярная диагностика 193 ния ДНК-типирования сначала берут часть биологического образца (пробу крови, сперму, кусочек кожи, волосы) и определяют, достаточно ли в нем интактной ДНК для последующего анализа. Затем ДНК подвергают эндонуклеазному расщеплению, полученные фрагменты разделяют в агарозном геле и переносят на най-лоновый фильтр. Проводят последовательную гибридизацию с четырьмя или пятью радиоактивно меченными зондами, каждый из которых распознает определенную последовательность ДНК (при этом перед гибридизацией со следующим зондом предыдущий полностью удаляют с мембраны). После каждой гибридизации с помощью радиоавтографии визуализируют полосы, отвечающие продуктам гибридизации зонда с рестрицированной ДНК, и строят «лестницу фрагментов» для всех образцов (рис. 9.7). Каждый этап (гибридизация и радиоавтография) длится от 10 до 14 сут, так что вся процедура может занять много недель и даже несколько месяцев. В качестве зондов обычно используют минисателлитные ДНК, многократно встречающиеся в геноме человека и состоящие из тандемно повторяющихся участков. Длина повторов варьирует от 9 до 40 п. н., а их число — от 10 до 30; при этом одни и те же минисателлитные последовательности у разных индивидов могут иметь разную длину. Зги различия возникают в результате увеличения или уменьшения числа тандемных повторов, по-видимому, в ходе репликации ДНК. Никаких биологических последствий такие вариации не имеют, поскольку минисателлитные ДНК не кодируют белков. Ребенок наследует одну минисателлитную последовательность от одного родителя, а другую — от другого. «ДНК-отпечаток" данного индивида представляет собой набор различающихся по длине фрагментов, соответствующих минисателлитным последовательностям его генома. Ввиду большого разнообразия этих повторов вероятность того, что в популяции найдется два человека с идентичными «ДНК-отпечатками", равна 105—108. Другими словами, характер расположения полос минисателлитных ДНК почти столь же индивидуален, как и отпечатки пальцев. Геномную дактилоскопию применяют также при установлении отцовства. Часть полос «ДНК-отпечатка» ребенка должна соответствовать полосам материнского «отпечатка», а часть — отцовского. Если ДНК в исследуемом образце недостаточно, но она не очень сильно разрушена, можно амплифицировать небольшие участки мини-сателлитной ДНК с помощью ПЦР, а затем провести их секвенирование; этот метод более
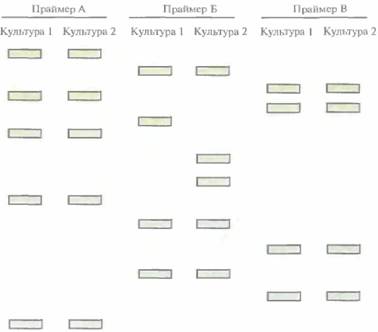
194 ГЛАВА 9 чувствителен, чем определение полиморфизма длины тандемных повторов. Использование полиморфных ДНК-маркеровМетод «ДНК-отпечатков» может оказаться полезным и при установлении различий между растительными культурами. Один из вариантов этого метода основан на использовании полиморфных ДНК-маркеров для амплификации случайных фрагментов {random amplified polymorphic DNA, RAPD). Для этого берут произвольные праймеры длиной 9—10 нуклеотидов, не содержащие палин-дромных последовательностей и имеющие ОС-содержание 50—80%, и добавляют их по отдельности к препаратам хромосомной ДНК растений. Каждая ПЦР инициируется одним праймером, который должен быть способен связываться с обеими цепями ДНК-мишени. Нуклеотидные последовательности всех олигонуклеотидов известны, но какой из них окажется эффективным инициатором ПЦР, неясно. Если праймер гибридизуется с обеими цепями ДНК-мишени в подходящей ориентации и сайты расположены на расстоянии от 100 до 3000 п. н. друг от друга, то фланкированный ими сегмент ДНК будет амплифицирован, а полученный фрагмент можно выделить с помощью гель-электрофореза и визуализировать окрашиванием. Число разных фрагментов ДНК, образующихся при амплификации, зависит от праймера и геномной ДНК. Для одних и тех же праймера и ДНК-мишени продукты амплификации будут каждый раз одинаковыми, а замена лишь одного нуклеотида в праймере приведет к полной смене набора получаемых фрагментов. Таким образом, используя один и тот же набор олигонуклеотидных праймеров, можно сравнивать RAPD-«ДНК-отпечатки» разных растительных культур, а следовательно, и сами культуры. Для выявления различий между двумя очень близкими сортами или культурами растений часто приходится использовать несколько произвольных праймеров с известной нуклеотидной последовательностью (рис. 9.8). Как и все другие молекулярные маркеры, RAPD можно применять для характеристики целых геномов, отдельных хромосом или генов. По сравнению с другими методами идентификации сложных ДНК метод RAPD обладает следующими преимуществами: 1) для всех видов растений можно использовать один и тот же (универсальный) набор олигонуклеотидных праймеров; 2) не нужно создавать геномные биб-
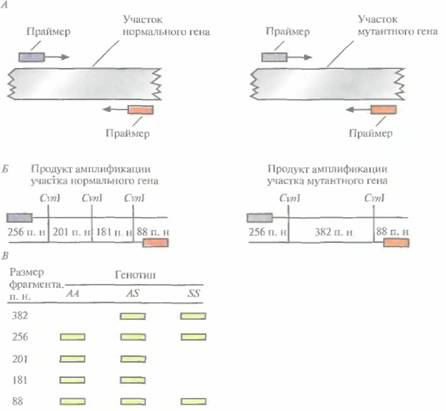
Молекулярная диагностика 195 лиотеки, использовать радиоактивные зонды, проводить гибридизацию, т. е. можно легко и быстро охарактеризовать большое количество образцов; 3) процесс можно автоматизировать. Кроме того, для проведения обычной ПЦР необходимо знать нуклеотидную последовательность искомого гена или его фрагмента — мишени для амплификации. В случае же RAPD амплифицируется любой участок генома, содержащий две комплементарные праймеp у последовательности, которые фланкируют сегмент ДНК длиной от 100 до 3000 п. н. С помощью метода RAPD удалось отличить друг от друга шесть инбредных линий кукурузы и показать, что ПЦР-продукты гибридов кукурузы представляют собой сочетание ПЦР-продуктов родительских инбредных линий. RAPD-маркеры использовали также для скрининга разных штаммов грибов Leptosphaeria maculans, вызывающих заболевание «черная ножка» у крестоцветных. Было установлено различие между невирулентным (не приводящим к развитию болезни) и вирулентным (вызывающим заболевание) штаммами. Молекулярная диагностика генетических заболеванийДиагностика специфических наследственных заболеваний человека на генетическом уровне дает ответ на вопрос, входят ли обследуемые индивидуумы или их потомки в группу повышенного генетического риска. ДНК-анализ можно использовать для выявления носителей генов наследственных заболеваний, а также для пренатальной и пресимптоматической диагностики серьезных генетических нарушений. Тесты на уровне ДНК позволяют безошибочно выявлять специфические мутации. Раньше для этого применялись биохимические методы, основанные на выявлении продукта анализируемого гена. ДНК-тесты не требуют экспрессии мутантного гена для его выявления, что позволяет разработать системы скрининга для всех моногенных заболеваний. Серповидноклеточная анемияСерповидно клеточная анемия генетическое заболевание, обусловленное заменой одного нуклеотида в кодоне, который соответствует шестой аминокислоте в ß-цепи молекулы гемоглобина. У индивидов, гомозиготных по мутантному гену (S/S), эритроциты имеют необычную серповидную форму; это связано с искажением конформации молекулы гемоглобина вследствие замены в ней валина на глутаминовую кислоту. Мутантный гемоглобин не может с достаточной эффективностью переносить кислород, и у таких больных развивается тяжелая анемия с прогрессирующим поражением сердца, легких, мозга, суставов и других органов. У индивидов, гетерозиготных по данному гену (A/S) (носителей генетического заболевания), эритроциты имеют нормальную форму, и симптомы заболевания проявляются лишь в экстремальных условиях (на большой высоте над уровнем моря либо при слишком высоких или низких температурах, когда снижается снабжение организма кислородом). Если оба родителя гетерозиготны (имеют генотип A/S), то вероятность того, что их ребенок будет гомозиготным по мутантному гену (S/S) (т. е. будет болен серповидноклеточной анемией), составляет 25%. Ген серповидноклеточной анемии с высокой частотой встречается среди афроамериканцев и их потомков, а также среди латиноамериканцев. В США проводят скрининг для выявления носителей гена серповидноклеточной анемии, которые могут передать этот ген своим потомкам. Рассмотрим один из используемых для этого тестов. Замена одного нуклеотида в ß-глобиновом гене, приводящая к серповидноклеточной анемии, сопровождается элиминацией сайта для рестрицирующей эндонуклеазы CvnI. Этот фермент узнает последовательность CCTNAGG и расщепляет молекулу ДНК между основаниями С и Т (N -любой из четырех нуклеотидов). В нормальном гене эта последовательность имеет вид CCTGAGG, а в гене серповидноклеточной анемии — ССТ-GTGG. На этом различии основывается ДНК-диагностика данного заболевания (рис. 9.9). Используя праймеры, фланкирующие сайт Cvn1, амплифицируют с помощью ПЦР небольшое количество тестируемой ДНК (рис. 9.9, А), Амплифицированный фрагмент обрабатывают Cvnl, продукты рестрикции разделяют с помощью гель-электрофореза и окрашивают их бромистым этидием. При наличии Сvn1-сайта на электрофореграмме появляется специфический набор полос (рис. 9.9, В), отличный от такового 196 ГЛАВА 9
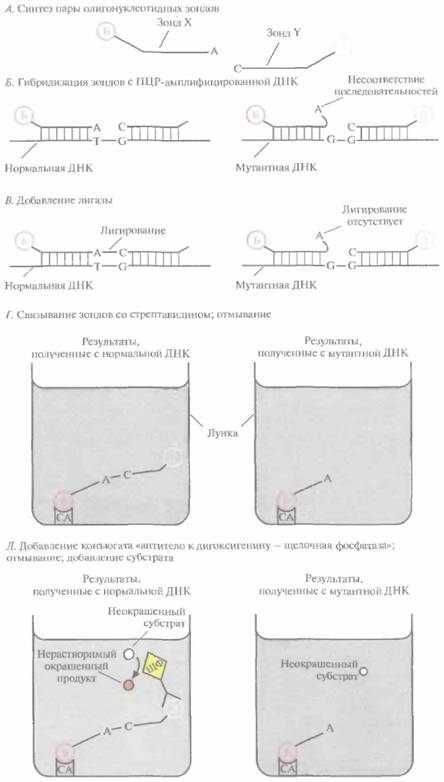
в отсутствие Cvnl-сайта. Описанным способом можно без труда и достаточно быстро установить генетический статус обследуемого, не проводя при этом процедуру гибридизации, Метод ПЦР/ЛОЗНе все генетические нарушения, приводящие к появлению дефектных генов, сопровождаются утратой или изменением сайтов рестрикции, поэтому для обнаружения однонуклеотидных замен применяют и другие подходы. В одном из них объединены ПЦР и метод, основанный на лигировании олигонуклеотидных зондов (ЛОЗ), ПЦР/ЛОЗ. Предположим, что в определенном сайте нормального гена (скажем, в 106-м положении) находится пара А-Т, а в том же сайте мутантного гена — G-C. Зная нуклеотидные последовательности, фланкирующие 106-й нуклеотид, можно синтезировать два коротких (20-нуклеотидных) фрагмента, прилегающих к данному сайту и комплементарных противоположным цепям (рис. 9.10). Основная особенность этой пары олигонуклеотидов состоит в том, что 3'-концевой нуклеотид одного из них (зонд X) комплементарен основанию, находящемуся в 106-м положении нормальной последовательности, а 5'-концевой нуклеотид второго (зонд Y) комплементарен нуклеотиду, примыкающему к 106-му нуклеотиду. При отжиге этих зондов с содержащей нормальную последовательность ДНК-мишенью (амплифицированной методом ПЦР) происходит их полная гибридизация, и при добавлении в реакционную смесь ДНК-лигазы зонды X и Y ковалентно сшиваются. Если же эти зонды отжигаются с мутантной ДНК, в которой произошла замена 106-го нуклеотида, то некомплементарный ему 3'-концевой нуклеотид зонда X не может образовать с ним пару. И хотя зонд Y по-прежнему гибридизуется полностью, ДНК-лигаза не может сшить зонды X и Y. Молекулярная диагностика 197
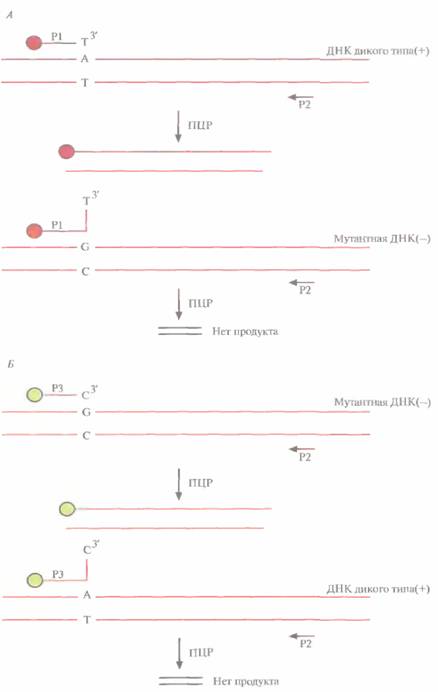
198 ГЛАВА 9 Можно синтезировать и другие олигонуклеотидные зонды, полностью соответствующие последовательности с мутантным 106-м нуклеотидом. При таком наборе зондов лигирование будет происходить в случае их отжига с мутантной ДНК-мишенью и не будет в случае отжига с нормальной мишенью. Таким образом, метод ПЦР/ЛОЗ различает две ситуации: лигирование зондов и отсутствие лигирования. Чтобы определить, произошло ли лигирование, 5'-конец зонда X метят биотипом, а 3'-конец зонда Y — дигоксигенином, низкомолекулярным соединением, связывающимся с соответствующим антителом. После гибридизации и лигирования проводят денатурацию ДНК для высвобождения гибридизовавшегося зонда и переносят смесь в небольшую пластиковую лунку, покрытую стрептавидином. Лунку промывают, чтобы удалить весь материал, кроме связавшегося со стрептавидином биотинилированного зонда. Затем добавляют в лунку антитела к дигоксигенину, предварительно соединенные со щелочной фосфатазой. После промывания, в ходе которого происходит удаление несвязанного конъюгата, добавляют бесцветный хромогенный субстрат. Окрашивание раствора в лунке свидетельствует о связывании антитела к дигоксигенину с зондом, меченным дигоксигенином, т. е. о том, что этот зонд был лигирован с зондом, меченным биотином. Если же окрашивания не происходит, значит лигирования не было. Располагая двумя парами зондов, можно установить генетический статус любого человека. Например, ДНК гетерозиготных носителей дает положительный ответ с обеими парами зондов, ДНК лиц, обладающих двумя копиями нормального гена, — только с тем набором зондов, который содержит нуклеотид, комплементарный нормальному сайту, и, наконец, ДНК индивидов с двумя измененными копиями гена — только с набором зондов, детектирующим мутантный сайт. Чтобы минимизировать необходимое для анализа количество исходной ДНК, перед гибридизацией участок ДНК-мишени, содержащий тестируемый сайт, амплифицируют с помощью ПЦР, ПЦР/ЛОЗ является быстрым, чувствительным и высокоспецифичным методом. Все его стадии роботизированы, что позволяет проводить до 1200 тестов в день. Более простым, хотя и менее чувствительным вариантом ПЦР/ЛОЗ является метод лигазной цепной реакции. Тестируемую ДНК смешивают с избытком двух индикаторных зондов, описанных выше, в присутствии термостабильной ДНК-лигазы. Проводят лигирование при 65 °С, затем повышают температуру до 94 °С, чтобы произошла денатурация образовавшихся гибридов зонд—ДНК-мишень, и вновь понижают температуру до 65 °С для гибридизации свободных нелигированных индикаторных ЛОЗ-зондов с ДНК-мишенью. Этот цикл повторяют 20 раз. Если индикаторные ЛОЗ-зонды полностью комплементарны ДНК-мишени, то лигирование будет происходить в каждом цикле, и после 20 циклов накопится достаточно продуктов лигирования («сшитых» зондов X и Y) для того, чтобы их можно было обнаружить с помощью электрофореза или ELISA. Если комплементарность неполная, то лигирование не произойдет и никаких продуктов зарегистрировано не будет. Генотипирование с использованием флуоресцентно меченных ПЦР-праймеровКолориметрическое генотипирование основано на применении ПЦР-праймеров, меченных различными флуоресцентными красителями. Чтобы различить мутантную ДНК и ДНК дикого типа, проводят ПЦР с двумя разными праймера-ми. Один из них (Р1) комплементарен ДНК дикого типа и на 5'-конце помечен родамином (красный цвет), другой (РЗ) комплементарен мутантной ДНК и на 5'-конце помечен флуо-ресиеином (зеленый цвет) (рис, 9.11). В обоих случаях амплификацию проводят в присутствии третьего, немеченного праймера (Р2), комплементарного противоположной цепи. Поскольку ПЦР может идти только в том случае, когда праймер полностью комплементарен ДНК-мишени, в присутствии в реакционной смеси всех трех праймеров будет амплифицироваться либо ДНК дикого типа, либо мутантная ДНК, либо обе они, в зависимости от ДНК-мишени, играющей роль матрицы. Если индивид гомозиготен по ДНК дикого типа, то после проведения ПЦР и удаления лишних праймеров будет наблюдаться флуоресценция красного цвета, если он гомозиготен по мутантной ДНК — зеленого, а если присутствуют и мутантная ДНК, и ДНК дикого Молекулярная диагностика 199 типа (т. е. индивид гетерозиготен) — желтого. Этот метод можно автоматизировать и адаптировать для любого однонуклеотидного сайта-мишени в любом гене с известной нуклеотидной последовательностью. Мутации в разных сайтах одного генаДалеко не все генетические заболевания обусловливаются одним специфическим изменением в гене. В большинстве случаев мутации происхо-
200 ГЛАВА 9 дят в разных сайтах в пределах одного гена, но приводят к одному генетическому заболеванию. В качестве примера можно привести ß-талассемию — наследственное заболевание, связанное с утратой активности ß-глобина. У гетерозиготных носителей при этом обычно наблюдается небольшая анемия. Индивиды же, гомозиготные по одному из как минимум восьми возможных мутантных сайтов, для поддержания жизни нуждаются в регулярном переливании крови и другом лечении. Поскольку мутация в любом из восьми специфических сайтов ß-глобинового гена может приводить к ß-талассемии, необходимо провести по крайней мере восемь разных тестов. Такая диагностика возможна, хотя и весьма дорогостояща. Поэтому для скрининга мутаций, возникающих в разных сайтах одного гена, была разработана стратегия ПЦР/гибридизация, основанная на проведении одной реакции. Для этого синтезируют набор специфических 20-нуклеотидных зондов, каждый из которых полностью комплементарен фрагменту гена-мишени, несущему известную мутацию. К 3'-концу каждого зонда присоединен гомополимер poly(dT) длиной
Микробиологическое производство лекарственных средств 201 примерно 400 нуклеотидов, с помощью которого ДНК-зонд связывается с заранее отмеченной точкой на найлоновом фильтре, а остальная его часть остается свободной и может гибридизоваться (рис. 9.12). Сегменты тестируемой ДНК, каждый из которых включает по одному из возможных мутационных сайтов, одновременно амплифицируют с помощью ПЦР, причем один пpaймер из каждой пары на 5'-конце помечен биотипом. Амплифицированные фрагменты ДНК-мишени гибридизуют с зондами, пришитыми к фильтру, в условиях, обеспечивающих гибридизацию только полностью комплементарных последовательностей. В гибридизационную смесь добавляют стрептавидин, связанный с щелочной фосфатазой (можно также использовать пероксидазу хрена или уреазу). После гибридизации промывают фильтр и добавляют неокрашенный субстрат. Если имеет место полное соответствие между амплифицированным сегментом ДНК-мишени и специфическим олигонуклеотидным зондом, то на фильтре появится цветная точка. На один и тот же фильтр можно нанести несколько точек, соответствующих целому ряду разных специфических олигонуклеотидных зондов. Проанализировав эту цветную мозаику, можно идентифицировать один из многих возможных сайтов мутации. ПерспективыМолекулярная диагностика — это быстро развивающееся направление. Хотя его основные принципы уже сформировались, технические детали отдельных тестов могут различаться. Для получения в достаточном количестве ДНК-мишени сейчас успешно применяют ПЦР. Использование ПЦР и специфических зондов существенно повышает чувствительность тестов и позволяет применять нерадиоактивные хромогенные, хемилюминесцентные и флуоресцентные системы регистрации. Во многих случаях для выявления мутации или экзогенной ДНК инфекционного агента в исследуемом образце достаточно провести ПЦР с последующим электрофоретическим разделением продуктов. Не вызывает сомнения, что с помощью ДНК-диагностики можно будет выявлять большинство, а возможно и все наиболее распространенные генетические и инфекционные заболевания, а также новообразования. ЗАКЛЮЧЕНИЕЛюбой эффективный диагностический тест должен быть: 1) высокоспецифичным в отношении молекулы-мишени; 2) достаточно чувствительным для выявления небольших количеств мишени; 3) достаточно простым, позволяющим без труда получать однозначные результаты. Существуют два типа методов молекулярной диагностики: один основан на сродстве антитела к конкретному антигену, другой — на идентификации специфических нуклеотидных последовательностей с помощью гибридизации или ПЦР. Наиболее распространенным иммунологическим методом является ELISA. Вкратце он состоит в следующем: 1) фиксация образца на твердой подложке; 2) добавление первого антитела, специфичного к антигену-мишени, и его связывание с антигеном-мишенью; 3) добавление конъюгата второе антитело-фермент, который присоединяется к первому антителу; 4) добавление неокрашенного субстрата, который под действием фермента, входящего в состав конъюгата, превращается в окрашенное соединение. Изменение цвета реакционной смеси свидетельствует о присутствии в образце молекулы-мишени. EL1SA применяется для обнаружения различных белков, идентификации вирусов и бактерий, а также определения низкомолекулярных соединений в широком спектре биологических образцов. Чтобы повысить специфичность первых антител, для диагностики часто используют моноклональные антитела. При этом для уменьшения стоимости прибегают к технике клонирования их фрагментов в Е. coliи получают комбинаторную библиотеку, а на ее основе — широкий спектр комбинаций Fv-фрагментов. Высокочувствительным и специфичным методом обнаружения нуклеотидных последовательностей в биологических образцах является гибридизация. Его использовали при разработке способов идентификации патогенных микроорганизмов в клинических образцах и различных микроорганизмов в окружающей среде. 202 ГЛАВА 9 ДНК-диагностика основывается на обнаружении известных нуклеотидных последовательностей; для этого синтезируют специфические праймеры и амплифицируют последовательность-мишень. Это позволяет использовать нерадиоактивные системы детекции (например, хемилюминесцентный метод) или регистрировать ПЦР-продукты методом гель-электрофореза. Кроме того, ПЦР-продукты можно пометить флуоресцентным красителем, присоединив его к 5'-концу праймера. В судебной медицине все более широкое применение находит метод геномной дактилоскопии, основанный на том, что ДНК каждого человека образует уникальный набор гибридизационных полос. При этом в качестве зондов обычно используют минисателлитные ДНК человека, которые не кодируют никаких белков и отличаются высокой вариабельностью. Для характеристики ДНК растений используют набор произвольных олигонуклеотидных праймеров, проводят ПЦР-амплифицикацию случайных фрагментов ДНК, осуществляют электрофорез и получают специфичный для каждого растения набор полос ДНК; данный подход носит название RAPD. Методы ДНК-диагностики применяют также для обнаружения точковых мутаций в данном гене. Один из подходов заключается в лигировании двух олигонуклеотидных праймеров. При несоответствии всего одного нуклеотида в месте стыковки гибридизовавшихся олигонуклеотидов лигирования не происходит. ЛИТЕРАТУРАBarany F. 1991. Single-nucleotide genetic disease detection using cloned thermostable ligase. Proc. 1991 Miami Bio/Technol. Winter Symp. 1:88. Barker R. H., L, Suebsaeng, W. Rooney, G.C. Alecrim, H. V. Dourado, D. F. With. 1986. Specific DNA probe for the diagnosis of Plasmodium falciparum malaria. Science 231: 1434-1436. Bugawan T L., R. K. Saiki, С. H. Levenson, R. M. Watson, H. A. Erfich. 1988. The use of non-radioactive oiïgonucleotide probes to analyze enzy-matically amplified DNA for prenatal diagnosis and forensic HIA typing. Bio/Technology 6:943-947. Carlson D. Р., С. Superko, J. Mackey, M. E. Gaskill, P. Hansen. 1990. Chemilumines-cent detection of nucleic acid hybridization. Focus 12:9-12, Caskey C. T. 1987. Disease diagnosis by recombinant DNA methods. Science 236: 1223-1229. Chehab F. F., Y. W. Kan. 1989. Detection of specific DNA sequences by fluorescence amplification: a color complementation assay, Proc. Natl. Acad. Set. USA 86: 9178-9182. Debenham P. G. 1992. Probing identity: the changing face of DNA finge [printing. Trends Biotechnol. 10:96-102. Erlich H. A., D. Gelfand, J. J. Sninsky. 1991. Recent advances in the polymerase chain reaction. Science 252: 1643-1651. Gillani 1. C. 1987. Non-radioactive probes for specific DNA sequences. Trends Biotechnol. 5: 332-334. Hartskeerl R. A., M. Y. L.. De Wit, P. R. Klatser. 1989. Polymerase chain reaction for the detection of Mycobacterium teprae. J. Gen. Microbiol, 135: 2357-2364. Jeffreys A. J., A. MacLeod, K. Tamaki, D. L. Neil, D. G. Monckton. 1991. Mini satellite repeat coding as a digital approach to DNA typing. Nature 354: 204-209. Kingsbury D. T. 1987. DNA probes in the diagnosis of genetic and infectious diseases. Trends Biotechnol. 5: 107-111. Klevan L., G. Gebeyehu. 1990. Biotinylated nucleotides for labeling and detecting DNA. Methods Enzymol. 184: 561-577. Kohler G., С. Milstcin, 1975. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256: 495—497. Kuppuswamy M. N., J. VV. Hoffmann, C. K. Kasper, S. G. Spitzer, S. L· Groce, S. P. Bajaj. 1991. Single nucleotide primer extensioin to detect genetic diseases: cAperimental application to hemophilia В (factor IX) and cystic fibrosis genes. Proc. Natl. Acad. Sei. USA 88:1143-1147. Mathews J. A., L. J. Kricka. 1988. Analytical strategies for the use of DNA probes. Anal. Biochem. 169: 1-25. Nickerson D. A., R. Kaiser, S. Lappin, J. Stewart, L Hood, U. Landegren. 1990. Automated DNA diagnostic using an ELlSA-based oligonucleotide ligation assay. Proc. Natl. Acad. Sei. USA 87: 8923-8927. ' Микробиологическое производство лекарственных средств 203 Persing D. H., T. F. Smith, F. C. Tenover, T. J. White (ed.). 1993. Diagnostic Molecular Microbiology: Principles and Applications. Ametican Society for Microbiology, Washington, D.C. Plikaytis В. В., R. H. Gelber, T. M. Shinnick. 1990. Rapid and sensitive detection of Mycohacterium leprae using a nested-prime r gene amplification assay. /. Clin. Microbiol. 28: 1913-1917. Pollard-Knigth D., A. C. Simmonds, A. P. Schaap, H. Akhavan, M. A. W. Brady. 1990. Nonradio-active DNA detection on Southern blots by en/y-matically triggered chemiluminescence. Anal. Biochem. 185: 353-358. Etafalski J. A., S. V. Tingey. 1993. Genetic diagnostics in plant breeding: RAPDs, microsatellites and machines. Trends Genet. 9: 275-279. Saiki R. K., P. S, Walsh, C. H. Levenson, H. A. Fjlich. 1989. Genetic analysis of amplified DNA with immobilized sequence-specific oligonucleotide probes. Proc. Nail. Acad. Sei. USA 86:6230-6234. Sayler G. S., A. C. Layton. 1990. Environmental application of nucleic acid hybridization. Anna. Rev, Microbiol. 44: 625-648. Tyagi S., F.R. Κramer. 1996. Molecular beacons: probes that fluorescence upon hybridization. Nat. Bwtechnol. 14: 303-308. Waldmann T. A. 1991. Monoclonal antibodies in diagnosis and therapy. Science 252: 1657—1662. Weiss J. B. 1995. DNA probes and PCR for diagnosis of parasitic infections. Clin. Microbiol. Rev. 8: 113-130. White T. J., N. Arnheim, H. A. Erlich. 1989. The polymerase chain reaction. Trends Genet. 5: 18;:.--L8S White T. J., R. Made], D. H. Persuig. 1992. The polymerase chain reaction: clinical applications. Adv. Clin, Chem. 29: 161-196. Winter G., C. Milstein. 1991. Man-made antibodies. Nature 349: 293-299. Vu K. F., A. Van Deynze, K. P. Pauls. 1993. Random amplified polymorphic DNA (RAPD) analysis, p. 287-301. In B.R. Click and J.E. Thompson (ed.). Methods in Plant Molecular Biology and Biotechnology. CRC Press, Boca Raton, Fla, КОНТРОЛЬНЫЕ ВОПРОСЫ1. Кратко опишите, как с помощью ПЦР можно выявить изменения в гене ß-глобина человека, приводящие к серповидноклеточной анемии. 2. Изложите принцип метода ПЦР/ЛОЗ. 3. Что такое метод EL1SA? 4. Опишите три способа нерадиоактивного мечения ДНК. Каковы преимущества нерадиоактивных методов детекции? 5. Перед вами стоит задача разработать простой, чувствительный и воспроизводимый тест для обнаружения содержащего двухцепочечную ДНК вируса, вызывающего летальную инфекцию у крупного рогатого скота. Поскольку эффективность лечения зависит от ранней и точной диагностики заболевания, необходимо использовать методы, позволяющие выявлять вирус при его минимальном содержании в организме инфицированного животного, еще до появления каких-либо симптомов заболевания. Кратко опишите и обоснуйте последовательность ваших действий. 6. Что означают чувствительность, специфичность и простота применительно к диагностическим тестам? 7. Как в настоящее время диагностируют болезнь Чагаса? Каким образом можно усовершенствовать существующую процедуру? 8. Почему использование флуоресцентных красителей облегчает обнаружение специфических нуклеотидных последовательностей? 9. Что такое зонд — «молекулярный маяк» и как он действует? 10. Что такое геномная дактилоскопия и как ее используют для характеристики следовых количеств ДНК в судебной медицине? 11. Что представляет собой метод RAPD и как его используют для выявления генетических вариантов растительных культур? | ||||||||||||||||||||||||||||||||||||||