хроматография-лекции. Хроматографические методы. Общая характеристика методов
 Скачать 6.82 Mb. Скачать 6.82 Mb.
|

|
Пламенно-ионизационный детектор не дает показаний для следующего ряда соединений: COS, CS2, H2S, NO, NO2, NH3, CO, CO2, H2O, SiCl4, SiHCl3, SiF4. В случае присутствия в анализируемой пробе указанных соединений чувствительность детектора к другим соединениям не изменяется. Пламенно-ионизационный детектор – типичный представитель классического потокового детектора. В заключение приведем основные преимущества и недостатки этого детектора. Преимущества:
Недостатки:
1.3.5.4. Детектор электронного захвата В основе функционирования детектора электронного захвата лежит то положение, что молекулы многих веществ способны реагировать со свободными электронами с образованием стабильных отрицательных молекулярных ионов. Принципиальная схема детектора электронного захвата приведена на рис. 43. 1 2 3 4 5 6 7 8 9 10 1-10 см/с 105 см/с Рис. 43. Схема детектора электронного захвата 1 катод; 2 радиоактивный источник; 3 молекулы газа-носителя; 4 положительные молекулярные ионы газа-носителя; 5 отрицательные молекулярные ионы определяемых соединений; 6 определяемые молекулы; 7 – свободные электроны; 8 анод; 9 подача газа-носителя; 10 зона ионизации молекул газа-носителя Радиоактивный источник (2) испускает 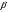 - - частицы, которые при столкновении с молекулами газа-носителя (как правило, азота) образуют свободные электроны и положительно заряженные молекулярные ионы- + N2 - - частицы, которые при столкновении с молекулами газа-носителя (как правило, азота) образуют свободные электроны и положительно заряженные молекулярные ионы- + N2 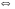 N2+ + e-. N2+ + e-.Под действием приложенного между электродами постоянного напряжения образовавшиеся в зоне ионизации свободные электроны движутся к аноду с очень высокой скоростью (порядка 105 см/c), несмотря на встречное движение потока газа-носителя. При этом в системе возникает электрический ток, который усиливается и регистрируется измерителем малых токов. Если в камеру детектора попадают соединения, способные захватывать электроны, то возможно протекание следующих процессов:
АВ + е-→ R-;
АВ + е-→ А- + В+ АВ + е-→ А- + В АВ + е-→ А + В-;
N2+ + e-→N2;
АВ- + N2+ →R + N2;
А- + N2+→ A + N2 B- + N2+→ B + N2. Все отмеченные процессы приводят к изменению концентрации заряженных частиц в камере детектора и будут оказывать влияние на величину тока в цепи. Если создать такие условия работы детектора, при которых имеет место только образование отрицательно заряженных молекулярных ионов анализируемого соединения, то величина уменьшения ионизационного тока будет зависеть только от концентрации анализируемого соединения в камере детектора. Уменьшение величины ионизационного тока обусловлено тем, что скорость движения отрицательно заряженных молекулярных ионов в камере детектора гораздо меньше скорости движения свободных электронов и составляет величину порядка 110 см/c. Встречный поток газа-носителя эту скорость еще дополнительно уменьшает, а на катоде в этом случае собираются только свободные электроны, концентрация которых зависит от концентрации молекул анализируемого соединения в камере детектора. Величина тока ионизации и концентрация присоединяющих электроны частиц связаны уравнением: 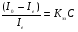 , (69) , (69)где Io и Ie ток ионизации в чистом газе-носителе и ток ионизации в присутствии присоединяющих электроны частиц соответственно; Кэз коэффициент захвата электронов данным соединением; С концентрация анализируемого соединения. Для сбора электронов в детекторе электронного захвата используется метод постоянного напряжения. Величина используемого напряжения может достигать 100 В. Чувствительность электронно-захватного детектора зависит от вероятности захвата молекулой исследуемого соединения электронов, которая в свою очередь зависит от присутствия в молекуле какого-либо захватывающего электроны атома или от структуры молекулы. Углерод и водород почти не имеют сродства к электронам, и углеводороды поэтому не захватывают свободных электронов. Исключение составляют высокомолекулярные ароматические соединения (антрацен), которые сильно захватывают электроны. Кислород и галогены легко захватывают электроны. В ряду галогенов степень поглощения электронов возрастает в ряду I > Br > Cl> F. В табл. 11 приведены относительные коэффициенты захвата электронов некоторыми классами соединений. При практическом использовании детектора электронного захвата необходимо учитывать следующие его особенности:
Т а б л и ц а 11 Величины относительных коэффициентов захвата электронов
Линейный диапазон детектирования детектора электронного захвата 102 – 104, предел обнаружения по линдану – 10-14 г/c. Детектор электронного захвата – потоковый детектор. Детектор электронного захвата применяют для анализа:
Следует отметить и основные недостатки детектора электронного захвата:
Для целей детектирования в газовой хроматографии применяются только так называемые закрытые источники, в которых исключена утечка радиоактивного вещества. Наиболее широко используются два типа источников: тритиевый и никелевый (63Ni). Существуют разнообразные конструкции ДЭЗ. Первый детектор, описанный Ловелоком (рис. 1.22, а), имел вид конденсатора с плоскопараллельными электродами, на одном из которых был размещен источник. Примером другой типичной конструкции является коаксиальный детектор (рис. 1.22, б), в котором один электрод выполнен в виде цилиндра с источником на внутренней поверхности, а другой – в виде стержня, расположенного на оси цилиндра; характеристики обоих типов детекторов довольно близки. 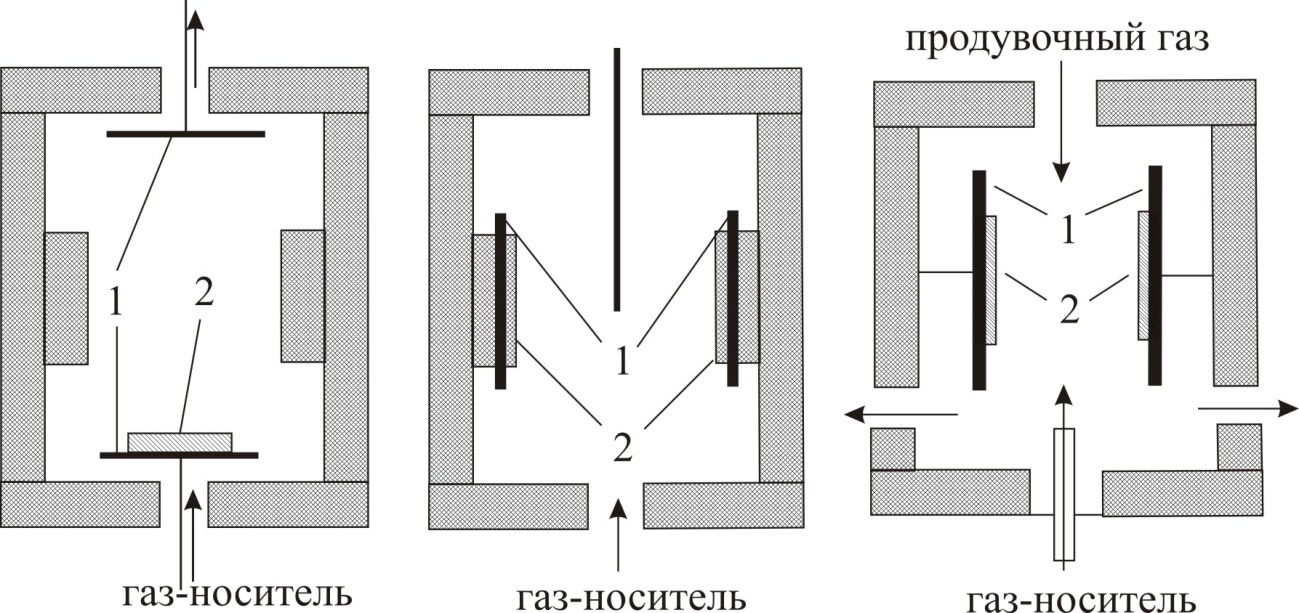 Несколько позднее были предложены (Грегори) более совершенные варианты конструкции (рис. 1.22, в), в которой зона ионизации продувочного газа конструктивно отделена от зоны захвата электронов молекулами пробы. Катод имеет форму цилиндра, на поверхность которого прикреплен радиоактивный источник. Продувочный газ обтекает катод, подвергаясь ионизации в зоне катода. Газ-носитель из колонки поступает через сетку анода, выполненного в виде стержня с осевым каналом. Эффективная зона захвата расположена в непосредственной близости от анода. Такая схема имеет ряд преимуществ, состоящих в том, что ионизируется только продувочный газ, а анализируемые вещества непосредственно не подвергаются действию радиации. Радиоактивный источник всегда находится в потоке чистого газа, и его загрязнения исключены. 1.3.5.5. Детектор ионизационно-резонансный Детектор ионизационно-резонансный является результатом совершенствования детектора электронного захвата. В детекторе электронного захвата используется метод постоянного питания, когда для сбора электронов применяется постоянное напряжение величиной до 100 В, что приводит к мешающему влиянию отмеченных выше процессов. Для устранения мешающих явлений, обусловленных использованием для сбора электронов постоянного напряжения, был предложен импульсный метод питания, который позволяет стандартизировать электронную температуру (приблизив ее к равновесной тепловой) и подвижность электронов, улучшить условия рекомбинации отрицательных и положительных ионов и уменьшить влияние пространственного заряда. Для сбора электронов применяются короткие импульсы потенциала с более длительными интервалами между ними. Импульсов амплитудой 50 В и длительностью 0.5 мкс обычно достаточно, чтобы собрать все присутствующие в детекторе электроны, и недостаточно, чтобы начался сбор на аноде отрицательно заряженных молекулярных ионов. При интервале между импульсами около 100 мс за счет рекомбинации и диффузии к стенкам детектора теряется не более 5 % электронов, выделенных источником излучения. За это время все электроны успевают собраться на коллекторе, и процессы захвата электронов молекулами анализируемых соединений идут в отсутствии электрического поля, т.е., когда электроны имеют, как и молекулы газа, практически тепловую энергию. 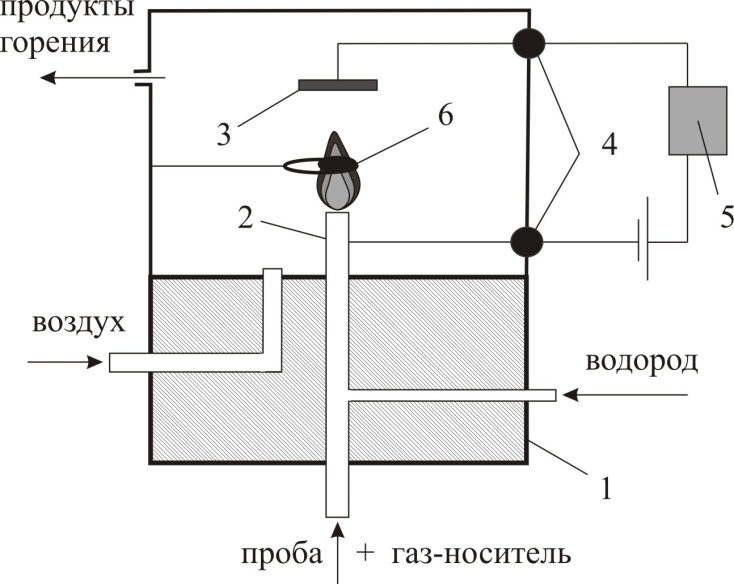 1.5.5.6. Термоионный детектор Название «термоионный детектор» применяется в настоящее время для целого ряда детектирующих систем, в которых ионизационный ток возникает благодаря термически генерированным ионам. В водородное пламя непрерывно поступает поток ионов щелочных металлов (Cs, Na, K). В присутствии этих ионов резко возрастает эффективность ионизации соединений, содержащих атомы некоторых элементов, в частности азота, фосфора, хлора и др. Поэтому термоионный детектор является селективным по отношению к таким соединениям. Различают два механизма возникновения таких ионов. Первый– поверхностная ионизация, при которой вещества с низким потенциалом ионизации могут ионизироваться на горячих поверхностях (например, на платине) с достаточно большой работой выхода, что наиболее типично для щелочных металлов. Такой детектор обычно называется детектором поверхностной ионизации (ПИ). Второй механизм состоит в том, что соли щелочных металлов (например, CsBr) термически испаряются в водородном пламени и таким образом в газовой фазе объема детектора устанавливается равновесие, например, CsBr + H+→ Cs+ + HBr, которое с увеличением температуры сдвигается в сторону увеличения диссоциации соли CsBr→ Cs + Br и протекания процесса ионизации Cs→ Cs+ + e,- что приводит к возникновению большого по величине тока ионизации. Одновременно в пламени протекают процессы диссоциации и ионизации разделяемых органических соединений, а также реакции рекомбинации: ОРГ →О + Р + Г диссоциация О →O+ + e- ионизация О+ + е-→ О рекомбинация Для фосфорсодержащих групп процесс ионизации Р→ Р+ + е- требует достаточно малых затрат энергии и приводит к созданию высокой концентрации ионов Р+ в потоке газа-носителя. Таким образом, оказывается, что процесс, обратный ионизации – процесс рекомбинации Р+ + е-→ Р протекает в присутствии как высокой концентрации электронов процесса ионизации соли щелочного металла, так и высокой концентрации ионов фосфора, а поэтому характеризуется высокой степенью вероятности и приводит к резкому уменьшению величины ионизационного тока. Следовательно, введение паров соли щелочного металла в водородное пламя приводит к проявлению термоионного эффекта в виде сильного увеличения чувствительности детектора к определенной группе органических соединений, например к фосфор- и азотсодержащим органическим соединениям. Изучение механизма работы детектора показало, что основные процессы происходят в пламени, а соль служит только источником атомов щелочного металла. Недостатками работы таких детекторов являются сильная зависимость основных характеристик детектора от расходов газов, трудность замены солевого резервуара, быстрое истощение запасов соли щелочного металла и соответственно низкое время непрерывной работы детектора без изменения его основных характеристик, загрязнение соли продуктами горения анализируемых веществ, приводящие к нестабильности работы детектора. Для устранения перечисленных недостатков была предложена новая конструкция более стабильного термоионного детектора, в которой рабочий объем камеры детектирования отделен от места образования паров соли щелочного металла. тефлоновый изолятор коллекторный электрод детектора ионизации диффузор воздуха изолятор горелка детектора ионизации корпус детектора наконечник из соли щелочного металла поджиг пламени продукт горения воздух водород коллекторный электрод термоионного детектора газ-носитель, водород горелка термоионного детектора Такой детектор получил название термоаэрозольного детектора и представляет собой комбинацию обычного пламенно-ионизационного детектора с генератором аэрозоля соли щелочного металла (рис. 44.). Рис. 44. Схема устройстватермоаэрозольного детектора Генератор аэрозоля состоит из трех частей:
из объема кварцевого держателя в поток инертного газа, необходимого для переноса паров соли в пламя детектора;
аэрозольные частицы (водяной холодильник);
Размеры, материал и размещение генератора аэрозоля относительно пламенно-ионизационного детектора выбирают таким образом, чтобы исключить распад и коагуляцию аэрозольных частиц до момента попадания их в пламя. На потенциальный электрод детектора подается положительное напряжение 100300 В, горелка и отрицательный электрод источника питания заземлены. Сигнал с коллекторного электрода подается на вход электрометрического усилителя и регистрируется потенциометром. Подача соли в виде аэрозольных частиц обеспечивает более стабильный поток щелочного металла. Соль щелочного металла испаряется в термостатируемой камере, температура которой с помощью терморегулятора поддерживается с погрешностью 0.1 оС. Температура нагрева соли около 500 оС. Образующийся пар потоком инертного газа (N2) выносится в охлаждаемую часть конденсатора и под действием поля с большим температурным градиентом охлаждается, переходит в состояние перенасыщения и затем в аэрозоль. Монодисперсность аэрозоля достигается путем разбавления его большим потоком инертного газа. Детектор может работать более 2000 часов без замены резервуара с солью при постоянстве чувствительности и силы фонового тока. Масса CsBr на держателе около 1 г. Перед установкой в генератор соль нагревается в течение 2030 часов при 400 оС для удаления летучих загрязнений, увеличивающих уровень шумов и дрейф детектора. Преимуществом данной конструкции термоаэрозольного детектора является незначительная зависимость его чувствительности, дрейфа нулевой линии и уровня шумов от изменений расходов газов (водорода, воздуха и газа-носителя) в довольно широкой области. Например, сигнал ДТА изменяется лишь на 1 % при изменении расхода водорода на 1 %, зависимость изменения чувствительности от изменения расхода газа-носителя еще меньше. Погрешность поддержания расхода водорода для обычного термоионного детектора должна быть не более 0.5 %, а газа-носителя 1.0 % от установленных значений. Поддержание такой стабильности расходов водорода и газа-носителя является достаточно трудной проблемой. Например, водородные генераторы непригодны для поддержания потока водорода с требуемой точностью. Однако для ДТА чувствительность практически не меняется при изменении расходов водорода в диапазоне 2337 см3/мин и воздуха 215360 см3/мин. При этом обеспечивается высокая стабильность работы детектора и воспроизводимость его показаний. Поскольку изменения температуры детектора и окружающей среды влияют на температуру соли щелочного металла и, следовательно, на стабильность работы детектора, необходимо температуру соли щелочного металла поддерживать постоянной с погрешностью не более 1 %. ДТИ отличаются чрезвычайно высокой чувствительностью и селективностью к соединениям, содержащим галогены и фосфор, а их чувствительность к хлору составляет около 210-6 %. Фосфорсодержащие соединения удается анализировать в экстрактах из овощей и фруктов на уровне 510-6 %. Установлено, что чувствительность ДТИ к соединениям, содержащим 6 атомов хлора, в 1000 раз меньше, чем чувствительность к соединениям, содержащим один атом фосфора. Установлено также, что, подбирая расходы газов и увеличивая расход водорода, можно повысить чувствительность ДТИ к азотсодержащим соединениям. Так, отношение чувствительности детектора к соединениям, содержащим Cl, As, N и Р, без учета влияния количества гетероатомов в молекуле и органической части молекулы, не содержащей этих атомов, примерно таково Cl : As : N : P = 1 : 20 : 100 : 1000. Минимально детектируемое количество при анализе фосфорсодержащих соединений составляет 510-14 г/с, а при анализе азотсодержащих – 510-13 г/с. Уровень шума при этом составляет около 1,510-13 А. Линейный диапазон детектирования 103. Такая высокая чувствительность детектора позволяет использовать его в приборах для охраны окружающей среды при анализе предельно допустимых концентраций вредных органических веществ. Так как ДТИ обладает наивысшей чувствительностью к фосфорсодержащим соединениям, наибольшее применение он нашел именно при анализе этих соединений. ДТИ применяется также для детектирования азотсодержащих соединений, причем правильный подбор экспериментальных параметров позволяет увеличить чувствительность детектора к этим соединениям на 23 порядка по сравнению с чувствительностью ДПИ. При определенных условиях ДТИ становится чувствительным и к галогенсодержащим соединениям, причем чувствительность детектора к ним примерно в 100600 раз ниже, чем к соединениям, содержащим фосфор, однако она на порядок выше чувствительности ДПИ. Детектор поверхностной ионизации применялся для высокочувствительного анализа хлоридов металлов. Предел детектирования на четыре порядка ниже, чем для детектора по теплопроводности и составляет, например, для SnCl4 и SiCl4 10-10 г/с. С помощью такого детектора можно определять неорганические хлориды при пределе детектирования 10-5 %. Отмеченные случаи аналитического применения ДТИ не исчерпывают всех его возможностей, однако, даже одна способность высокочувствительного детектирования фосфорсодержащих пестицидов оправдывает тот большой интерес, который проявляется к ДТИ. 1.3.5.7. Гелий-ионизационный детектор был разработан в 1950-е годы. Принцип действия ГИД основан на том, что ионизация инертного газа увеличивается, если при постоянном уровне облучения в него добавляют посторонний газ. Если в камере детектора имеется источник -частиц, например тритий, то при наличии поля, создаваемого высоким напряжением, гелий возбуждается и его атомы становятся метастабильными. Механизм процесса основан, вероятнее всего, на переносе энергии от метастабильного гелия к другим атомам и молекулам. Сначала образуются заряды с постоянной скоростью: А → А+ + е− Освободившиеся электроны малых энергий разгоняются сильным полем и при соударениях с атомами газа-носителя сообщают им энергию, переводящую их в возбужденное (метастабильное) состояние: А + е−→ А* + е− Полный сбор электронов и ионов, возникающих в результате первичной ионизации газа-носителя, создает фоновый ток детектора. Вероятность перехода возбужденных атомов Ar или Не в первоначальное энергетическое состояние значительно увеличивается при введении в детектор веществ, имеющих близкие или меньшие потенциалы ионизации (энергию отрыва электрона), чем энергия возбужденного состояния: А* + М → А + М+ + е− Образующиеся в результате реакции вторичной ионизации заряды создают дополнительный ток, являющийся сигналом детектора на введенное количество вещества. Так как энергия возбуждения метастабильного гелия (19,6 эВ) и аргона (11,6 эВ) больше, чем потенциал ионизации всех других частиц, за исключением неона (21 эВ), поэтому другие компоненты могут ионизироваться. ГИД используется главным образом в том случае, если необходимо обнаружить следы посторонних газов. ГИД является универсальным детектором. 1.3.5.8. Атомно-эмиссионный детектор (АЭД) В течение многих лет исследователи пытались использовать атомно-эмиссионную спектроскопию в газовой хроматографии. Ее применение дает возможность определять элементы непосредственно в элюате, поступающем из колонки. Возбуждающие атомы излучают свет с характерной длиной волны. В атомно-эмиссионном детекторе проба переводится в атомарное состояние, а образовавшиеся атомы переходят в возбужденное состояние. Для этого необходима значительная энергия, которая имеется в плазме, индуцированной микроволновым излучением. Переход возбужденных атомов в состояние с более низкой энергией сопровождается излучением света. Длина волны возникающего излучения измеряется спектрофотометром. А + энергия → А* А* → А + энергия или А + фотон → А+* + е− А+* → А+ + фотон где звездочкой отмечены на схеме частицы, находящиеся в возбужденном состоянии. 1.3.5.9.Фотоионизационный детектор (ДФИ) Детектор был предложен в 1968 г., имел нестабильные характеристики и почти не применялся. В конце 70-х начале 80-х годов началась новая эра в развитии ДФИ, связанная, главным образом, с его применением для анализа примесей в воздухе. Новые конструкции детектора имеют чувствительность и линейность на уровне или выше тех же параметров ДПИ, причем в качестве газа-носителя можно использовать воздух. Детектор применяют в портативных и автономных газовых хроматографах, специально разработанных для целей охраны окружающей среды. Проведено изучение работы ДФИ с различными газами-носи телями, определены его линейности и чувствительности для большого круга органических веществ. Испробовано применение ДФИ с капиллярными колонками. В качестве источника возбуждения использован лазер, с помощью которого исследован механизм двухфотонной фотоионизации для анализа полиатомных ароматических соединений. Принцип работы ДФИ состоит в следующем: фотоны от ультрафиолетовой (УФ) лампы попадают в ионизационную камеру, через которую непрерывно проходит газ-носитель, выбранный таким образом, чтобы его потенциал ионизации Ip был значительно выше энергии фотонов. В этом случае газ-носитель не ионизируется, в то время как попадание в ионизационную камеру анализируемого вещества вызывает появление фотоионизациойного тока, пропорционального концентрации этого вещества. Диапазон детектируемых соединений ограничен «сверху» – детектируются все соединения, в том числе и неорганические, для которых потенциал ионизации меньше энергии фотонов. Различные УФ-лампы могут обеспечить разную селективность ДФИ к различным соединениям за счет сведения сигнала к некоторым из них до минимума. В этом случае можно определять даже неразделенные хроматографические пики. Однако такого рода селективность ограничена выбором источников излучения, что в первую очередь связано с отсутствием материалов, пропускающих свет более коротковолновый, чем резонансное излучение аргона: Коротковолновая граница пропускания для ДФИ находится ниже 11,7 э В.  Теоретические вычисления чувствительности ДФИ не дают достоверных результатов. Однако практически установлено, что ДФИ в среднем и в зависимости от типа соединения в 10–30 раз более чувствителен и имеет в 10 раз больший линейный диапазон детектирования, чем ДПИ. Наряду с этим использование воздуха в качестве, газа-носителя и отсутствие пламени дают ДФИ неоспоримые преимущества по сравнению с ДПИ. Принципиальная схема ДФИ приведена на рис. 11.24. Свет от УФ-лампы 3 через окно 4 из MgF2 попадает в ионизационную камеру 1с потенциальным 5 и измерительными 2 электродами.. Через трубку, являющуюся потенциальным электродом 5, в камеру из хроматографической колонки поступаем газ-носитель. В качестве источника фотоионизации применена криптоновая УФ-лампа 3 тлеющего разряда типа. Ионизационная камера изготовлена из высокоомной керамйки с электродами из нержавеющей стали. Электроды и окно приклеены к керамическому корпусу специальным клеем. Максимальная рабочая температура такого детектора около 200 °С. При разработке детектора основные трудности связаны с технологией его изготовления, в том числе герметизацией УФ-лампы, окна из MgF2 и ионизационной камеры, выбором формы и материалов электродов при минимальном размере камеры и др. Одним из недостатков ДФИ является возможность загрязнения окна из MgF2 компонентами газа-носителя, пробы и неподвижной фазы. Загрязнение приводит к уменьшению потока фотонов и к значительной потере чувствительности. Фирма «Fotovak» (США) выпускает портативный газовый хроматограф с ДФИ для определения следов органических соединений в атмосфере. С помощью прибора можно определять органические вещества в 1 см пробы воздуха в количестве 0,1 млрд.-1. Преимущества прибора определяются высокостабильным источником фотонов с энергией да 11 эВ, который питается от высокочастотного генератора. Характерной особенностью прибора является возможность его применения при температуре окружающей среды, поэтому основные детали детектора изготовлены из фторопласта. Для идентификации многокомпонентных смесей может быть применен набор фотоионизационных детекторов с различными УФ-лампами. Соотношение между сигналами ДФИ, например с УФ-лампами на 9,5 эВ и 11,7 эВ, позволяет получить дополнительную информацию о природе анализируемых веществ. При работе с ДФИ в режиме ДЭЗ в качестве газа-носителя используют азот с примесью легко ионизируемого с помощью УФ-лампы органического вещества. Образовавшиеся электроны собираются на аноде под влиянием электрического поля и ддют фоновый ток, который уменьшается при захвате электронов электроотрицательными анализируемыми веществами. С помощью крана потоки газов-носителей переключаются таким образом, что детектор может последовательно работать в режимах ДФИ и ДЭЗ. Газом-носителем для режима ДЭЗ может служить азот с добавлением паров три-н-пропиламина или нафталина. Фоновый ток зависит от природы и количества добавок, интенсивности УФ-лампы и чистоты окна из MgF2. Для получения фонового тока 1-10-8 А поток нафталина должен составлять около 1 мкг/мин. При этих условиях можно анализировать антрацен на уровне 500 пг. Поляризационное напряжение составляло около 200 В. Линейный диапазон для линдана и гептахлора около 103. Интерес к ДФИ постоянно повышается и многие фирмы пла нируют ввести его в состав своих хроматографов в качестве одного из основных детекторов. 1.3.5.10. Редокс-хемилюминесцентный детектор (РХД) Этот вид детекторов был разработан в конце 1970-х годов для количественного анализа азота, водорода и соединений серы в воде или воздухе. Обычно для определения используется реакция азота с озоном. С помощью РХД можно анализировать следующие классы соединений: спирты, альдегиды, кетоны, фенолы, олефины, ароматические углеводороды, амины, тиолы, сульфиды и фосфонаты. РХД хорошо сочетается с ДИП, так как многие соединения, не дающие сигнала в детекторе ДИП, реагируют как восстановители и тем самым способны регистрироваться детектором РХД. 1.3.5.11. Инфракрасные детекторы (ИКД) Инфракрасная спектроскопия широко применяется в химическом анализе и в сочетании с газовой хроматографией. Методом ИК-спектроскопии с преобразованием Фурье (ИКПФ) проводят анализ элюируемых соединений с высокой скоростью и чувствительностью. Полученный при этом ИК-спектр поглощения можно рассматривать как индивидуальную характеристику соединения и использовать для его идентификации. 1.3.5.12 Масс-селективный детектор (МСД) Уже давно масс-спектрометр рассматривается как отличный детектор для газовой хроматографии. Полученные с его помощью спектры, подобно ИКД, дают такую информацию о качественном составе пробы, какую не могут дать иные газохроматографические детекторы. Различие между МСД и ИКД состоит в том, что первый обладает большей чувствительностью по сравнению с ИКД, кроме того, он разрушает пробу, дает информацию о массе, а не о функциональных группах и различает скорее гомологи, чем изомеры. При бомбардировке электронами молекул в газообразном состоянии связи в молекулах разрываются и образуют ионы. Вид и количество образующихся фрагментов характерны для данной молекулы. При наложении магнитного поля положительно заряженные частицы ускоряются и движутся по изогнутым кривым, радиус кривизны которых пропорционален корню квадратному из массы иона. При некотором постоянном магнитном поле поток ионов, содержащий ионы с идентичным масса/заряд, попадает на коллектор. Здесь при разряде ионов возникает ток, пропорциональный относительному количеству ионов с соответствующей массой. Изменением магнитного поля постепенно переводят на коллектор потоки ионов с другим соотношением масса/заряд. Ток коллектора записывается и дает масс-спектрограмму. В квадрупольном масс-спектрометре разделение по массе достигается иным образом. Между четырьмя постоянными магнитами образуется высокочастотное электрическое поле. Когда пучок ионов попадает в это поле, только ионы с определенным отношением масса/заряд имеют стабильную траекторию и попадают на детектор (коллектор). Детектирование пучков с различным отношением масса/заряд проводят варьированием электрического поля. 3.1. Варианты метода газовой хроматографии При классификации вариантов методов газовой хроматографии предполагается, что подвижная фаза (газ-носитель) абсолютно инертна к неподвижной фазе и разделяемым соединениям. Таким образом, классификация вариантов основывается только на особенностях неподвижной фазы. В качестве неподвижной фазы в газовой хроматографии используется или твердый адсорбент, или жидкость, нанесенная в виде тонкой пленки на адсорбционно инертный твердый носитель. В соответствии с типом используемых неподвижных фаз газохроматографические методы подразделяются на газо-адсорбционный и газо-жидкостный. Разделение компонентов анализируемой смеси в газо-адсорбционном варианте основано на различии разделяемых веществ в величинах адсорбции на поверхности адсорбента, а в случае газожидкостной хроматографии на различии в растворимости компонентов анализируемой смеси в неподвижной жидкой фазе. В том случае, если используемый твердый носитель неподвижной жидкой фазы проявляет адсорбционные свойства, реализуется промежуточный вариант газовой хроматографии – газо-жидко-твердофазная хроматография. Каждый из вариантов характеризуется своими положительными чертами и недостатками, которые обязательно следует учитывать при выборе оптимального метода разделения каждой конкретной смеси. Для получения достоверных результатов анализа необходимо подобрать оптимальные условия хроматографирования, к числу которых прежде всего следует отнести выбор температуры колонки, адсорбента, газа-носителя, его скорости, количества вводимой пробы, и др. 1.5.1 Газоадсорбционная хроматография. Особенность метода газоадсорбционной хроматографии состоит в том, что в качестве неподвижной фазы применяют адсорбенты с высокой удельной поверхностью (10–1000 м2/г) и распределение веществ между неподвижной и подвижной фазами определяется процессом адсорбции молекул разделяемых веществ из газовой фазы и их концентрированием на поверхности раздела твердой и газовой фаз за счет межмолекулярных взаимодействий. При осуществлении газоадсорбционной хроматографии первостепенное значение имеет правильный выбор адсорбента. Адсорбент должен обладать следующими свойствами: достаточной селективностью; химической и каталитической инертностью; изотермой адсорбции, близкой к линейной; достаточной механической прочностью. Селективность адсорбента определяется в первую очередь силами взаимодействия адсорбата с поверхностью адсорбента. В общем виде различают две группы сил: физические и химические, хотя между ними имеются и переходные моменты. При физической адсорбции взаимодействие разделяемых молекул с поверхностью адсорбента осуществляется за счет ориентационных, индукционных и дисперсионных сил, называемых в совокупности ван-дер-ваальсовыми. Силы полухимического взаимодействия – это прежде всего водородная связь и образование комплексов переноса заряда. И, наконец, хемосорбция протекающая за счет образования прочной химической связи между молекулами разделяемых веществ и адсорбентом. Силы дисперсионного взаимодействия Энергия дисперсионного взаимодействия двух сферических частиц описывается уравнением Лондона: 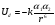 , (84) , (84)где k коэффициент пропорциональности, зависящий от потенциала ионизации частиц; 1, 2 поляризуемость частиц; r расстояние между частицами. Поскольку каждая частица обладает определенной поляризуемостью, дисперсионные силы проявляются при взаимодействии любых частиц. В растворах органических соединений дисперсионные силы вносят основный вклад в энергию межмолекулярного взаимодействия, а для неполярных молекул только они обуславливают меж- молекулярные взаимодействия. Поляризуемость частиц в первом приближении можно выразить через молекулярную рефракцию RM соотношением: 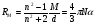 , (85) , (85)в котором n – показатель преломления исследуемого соединения; М – его молярная масса; d - плотность;N – число Авогадро. Минимально возможные расстояния между частицами определяются как сумма ван-дер-ваальсовых радиусов этих частиц. Однако достаточно точное вычисление энергии дисперсионных сил даже для простейших систем осложнено тем, что определение реальных расстояний между частицами существенно затруднено. Выходом из создавшегося положения может быть использование правила аддитивности для расчета дисперсионного взаимодействия молекул как суммы инкрементов отдельных атомов, входящих в состав этих молекул. Каждый атом или атомная группа обладают легко находимой по справочнику атомной рефракцией. Однако атомы и атомные группы экранируются внутри молекулы ближайшими частицами. Вследствие такого экранирования лишь часть поверхности атомной группы взаимодействует с окружающей средой. Эту часть можно определить для любой группы атомов или отдельных атомов, что дает возможность рассчитать для каждой группы коэффициент внутримолекулярного экранирования в зависимости от размеров ближайших частиц. Из уравнения (84) видно, что энергия дисперсионного взаимодействия уменьшается пропорционально шестой степени расстояния между частицами и поэтому без больших ошибок можно ограничиться рассмотрением взаимодействия между двумя наиболее близко расположенными атомными группами сорбата и неподвижной фазы. В этом случае соблюдаются основные условия использования уравнения. |