хроматография-лекции. Хроматографические методы. Общая характеристика методов
 Скачать 6.82 Mb. Скачать 6.82 Mb.
|

|
Адсорбенты с большим содержанием кремниевой кислоты Основными представителями этой группы адсорбентов являются: силикагель, пористые стекла, цеолитовые молекулярные сита. Силикагель представляет собой аморфный продукт конденсации поликремниевых кислот и характеризуется очень высокой величиной удельной поверхности. На поверхности силикагеля находятся силанольные группы  Si-OH, проявляющие кислотные свойства, а также силоксановые группы Si-O-Si , являющиеся донорами электронов. Si-OH, проявляющие кислотные свойства, а также силоксановые группы Si-O-Si , являющиеся донорами электронов.На 1 нм2 поверхности силикагеля приходится от 4 до 8 геминальных, вицинальных и изолированных силанольных групп: HO OH HO OH OH Si O Si Si Si геминальные вицинальные изолированные В большинстве случаев силикагель во влажном состоянии получают нейтрализацией растворов силиката натрия минеральными кислотами. Изменением рН можно регулировать диаметр пор образующегося геля. После промывки и нагревания образуется твердый пористый силикагель, структуру пор которого можно модифицировать гидротермальной или химической обработкой кислотами. Высокодисперсные образцы силикагеля с малой плотностью назы-вают аэрогелями. Силикагель можно также получать пирогенным методом: так, например, получают аэросил из хлорида кремния. Высокая концентрация свободных гидроксильных групп с частич-но протонированным водородом является причиной того, что силикагель специфически адсорбирует молекулы с высокой электронной плотностью (спирты, эфиры, кетоны и амины), а также неполярные молекулы с поляризующимися -связями (ароматические соединения, олефины). Адсорбционная активность силикагеля зависит от содержания в нем воды и снижается по нелинейному закону при увеличении ее количества. На поверхности гидратированного силикагеля в обычных условиях адсорбирован полимолекулярный слой воды. Третий от поверхности силикагеля – слой слабо адсорбированной воды, обратимое ее удаление происходит от комнатной температуры до 70 оС. Этот слой воды удаляется сухим растворителем. O H ОSiOH … OH … OH … OH O H H носитель Второй слой также слабо адсорбированной воды полностью удаляется при 120 оС, максимум удаления при 100 оС. Процесс является обратимым. Этот слой также удаляется сухим растворителем. Первый слой – слой сильно адсорбированной за счет водородной связи воды. Удаление этого слоя начинается при 200 оС и завершается при 650 оС. Процесс является обратимым, этот слой не удаляется сухим растворителем. В процессе нагревания силикагеля силанольные группы SiOH теряют воду и превращаются в силоксановые SiOSi. Потеря воды – процесс необратимый; начинается при 450 оС и завершается при температуре 1100 оС.В хроматографии силикагель применяется как в неактивном (полностью гидратированный), так и активном состоянии (безводный). В активном состоянии его используют для разделения смесей углеводородов, в неактивном состоянии – для разделения смесей полярных соединений. Основными преимуществами силикагеля являются:
Модифицирование поверхности силикагеля заключается в замещении силанольных групп на другие функциональные группы. Это позволяет наряду с устранением мешающих активных центров и повышением однородности поверхности адсорбента оказывать направленное воздействие на величину полярности этой поверхности. С другой стороны, появляется возможность получения неподвижных жидких фаз, закрепленных на носителе и используемых в газо-жидкостной хроматографии и в высокоэффективной жидкостной хроматографии. Пористые стекла. Нагреванием натрийборосиликатных стекол (обычный состав – 70 % SiO2, 23 % B2O3, 7 % Na2O) при 1400 оС получают гомогенный расплав, распадающийся при 500600 оС на две фазы, одна из которых состоит из практически чистого диоксида кремния, а вторая из бората натрия, как бы пронизывающего скелет диоксида кремния. После охлаждения боратно-натриевую фазу вымывают разбавленными кислотами и получают поликремниевый скелет. Поскольку структуру пористых стекол можно регулировать, меняя состав исходного стекла и условия процесса, этим методом получают пористые стекла с воспроизводимым диаметром пор с размером от диаметра молекулы до нескольких нанометров, причем с абсолютно одинаковым размером пор как на поверхности, так и внутри зерен. Следует иметь в виду, что стекла с внутренними порами имеют существенный недостаток, мешающий экспресс-анализу: процесс проникновения разделяемых молекул внутрь зерна протекает сравнительно медленно, что замедляет массообмен. Стекла с поверхностными порами, которые можно получить целенаправленным выщелачиванием раствором соляной кислоты, не имеют этого недостатка. Цеолитовые молекулярные сита. Природные и синтетические цеолиты представляют собой кристаллические алюмосиликаты с трехмерной структурой из тетраэдров SiO4 и AlO4, объединенных в те или иные полиэдры. Общая формула цеолитов следующая: M2/nO, Al2O3, xSiO2, yH2O, где М щелочной или щелочноземельный металл; n степень его окисления. Наиболее распространены цеолиты формулы: Na2O, СаО, Al2O3, xSiO2, yH2O. Цеолиты относятся к группе микропористых скелетных силикатов с большой внутренней удельной поверхностью (700800 м2/г). Эта поверхность образована ионами, которые несут значительные положительные и отрицательные заряды и придают ей гидрофильность. Характерной особенностью цеолитов является строго регулярная система пор – набор больших одинаковых пустот, связанных между собой однородными микропорами с шириной окон порядка диаметра молекулы. Заполняющая пустоты гидратная вода слабо связана с решеткой и удаляется при нагревании без заметного изменения структуры цеолита. Ввиду многообразия комбинаций катионов, возможности изменения отношения Si/Al, а также различных допустимых вариантов построений каркаса, класс цеолитов весьма велик и разнообразен. В цеолитах марки А трехмерные структурные единицы соединяются через четырехчленные кислородные кольца, в цеолитах Х и Y они соединяются через шестичленные кислородные кольца. Для цеолитов Х характерно отношение Si/Al от 1.0 до 4.0, а для цеолитов Y – от 2 до 3. Стенки пор образованы атомами кислорода и входные окна в поры могут в различной степени блокироваться катионами. Меняя катионы, можно направленно изменять величину диаметра пор. В газовой хроматографии наиболее часто применяются молекулярные сита марок 5А, 10Х и 13Х со средними геометрическими размерами пор 5, 7 и 9 ангстрем. Оксид алюминия Оксид алюминия является весьма термостойким и механически прочным адсорбентом; его удельная поверхность составляет около 200 м2/г. Из-за наличия кислотных и основных (по Льюису) центров его поверхность имеет гетерополярный характер. Адсорбент существует в нескольких полиморфных модификациях, из которых наиболее известными являются , и формы. Получают оксид алюминия обезвоживанием гидроксида алюминия: например, форма образуется при нагревании гидроксида алюминия до температуры 600 1000 о С. Состав активного в адсорбционном отношении оксида алюминия не соответствует строго формуле Al2O3; он содержит также гидроксильные группы, образующие активные центры наряду с уже отмеченными донорными и акцепторными центрами. Кроме приведенных недостатков, связанных с гетерогенным распределением пор и неоднородностью поверхности, оксид алюминия гигроскопичен, и, следовательно, его активность зависит от содержания воды на его поверхности. Поэтому перед применением в газовой хроматографии рекомендуется проводить предварительную обработку оксида алюминия. Добавление точно известного количества воды к заранее высушенному Al2O3 позволяет сократить “хвосты” пиков на хроматограммах и снизить время удерживания. Минимальная полярность адсорбента наблюдается при мономолекулярном покрытии поверхности водой. Органические сорбенты Наибольшее применение получили пористые сополимеры стирола и дивинилбензола. Эти материалы получают суспензионной полимеризацией мономерных винильных производных (стирола, этилбензола), к которым добавлено некоторое количество дивинилбензола для образования поперечных связей (сшивки). Меняя исходные компоненты и их соотношение, а также условия реакции полимеризации, можно направленно модифицировать не только удельную поверхность и диаметр пор, но и полярность материала. Особенно важным свойством этих полимеров является их гидрофобность, обусловленная отсутствием гидроксильных групп. Малое сродство к соединениям, содержащим гидроксильные группы, имеет следствием относительно малое время удерживания и симметричные пики воды, спиртов, гликолей, карбоновых кислот, аминов. Такие важные для газохроматографического применения свойства пористых полимеров, как высокая химическая и геометрическая гомогенность, механическая прочность, большая емкость вследствие большого объема пор, гидрофобность, а также сферическая форма частиц способствовали тому, что эти неподвижные фазы в короткое время получили широкое распространение и в настоящее время специально для газовой хроматографии выпускается большой набор подобных полимеров. 12.5. ПРИЛОЖЕНИЕ ТЕОРИИ АДСОРБЦИИ К ГАЗОВОЙ ХРОМАТОГРАФИИ Эффективность разделения веществ в газо-адсорбционной хроматографии определяется закономерностями процессов адсорбции. В этой связи целесообразно рассмотреть основные положения теории адсорбции с точки зрения их приложения к газовой хроматографии. 12.6. ОСНОВНЫЕ ПРЕИМУЩЕСТВА И НЕДОСТАТКИ ГАЗО-АДСОРБЦИОННОЙ ХРОМАТОГРАФИИ В заключение рассмотрения особенностей варианта газо-адсорбционной хроматографии отметим его основные положительные черты и недостатки. Положительным для газа-адсорбционного варианта является:
К недостаткам метода следует отнести:
1.5.2. Применение газо-адсорбционной хроматографии для решения практических задач контроля качества продукции. Газо-адсорбционная хроматография наиболее пригодна для анализа легких газов, к числу которых относится водород, азот, кислород, инертные газы, метан, оксид и диоксид углерода, оксиды азота и др. Газоадсорбционная хроматография позволяет разделять смеси изотопов Все эти вещества не детектируются ионизационными детекторами. Поэтому их анализ проводят с использованием катарометров или специальных высокочувствительных детекторов. Для разделения смесей указанных газов, а также изотопов необходимы сильные адсорбенты, обладающие специфическими свойствами. К ним относятся активированные угли, некоторые силикагели и алюмогели, а также молекулярные сита и пористые полимерные адсорбенты. Наиболее часто легкие газы разделяют при низких температурах порядка 70оС и ниже. 1.5.3. Газо-жидкостная хроматография. Газожидкостная хроматография на практике применяется гораздо чаще, чем газоадсорбционная. Это связано с тем, что набор веществ, которые могут быть использованы в качестве неподвижных жидких фаз, гораздо шире набора имеющихся активных адсорбентов. Эти неподвижные жидкие фазы имеют более широкий диапазон концентраций с линейной функцией изотермы распределения, что дает возможность работать с большими пробами и, следовательно, обеспечивает более высокую чувствительность анализа, а также позволяет с меньшими усилиями добиваться высокоэффективной и воспроизводимой работы колонок. В основе распределительной газожидкостной хроматографии лежит различие растворимости разделяемых веществ в выбранном неподвижном растворителе в хроматографической колонке или, точнее, различие коэффициентов их распределения между неподвижной жидкой фазой (НЖФ), служащей растворителем, и подвижной газовой фазой (ПГФ), которой является газ-носитель. Чем выше коэффициент распределения вещества в газо-жидкостной колонке, тем больше его удерживаемый объем и тем дольше вещество задерживается в колонке. Разделительная способность газожидкостной колонки зависит: – от правильности выбора носителя НЖФ; от правильности выбора НЖФ, т. е. такой, которая была бы наиболее селективной по отношению к разделяемым компонентам смеси; правильности выбора условий разделения (размеров колонки, ее температуры, скорости подачи газа-носителя, количества анализируемой пробы, вводимой в колонку). 1.5.4. Характеристики носителя неподвижной жидкой фазы и их влияние на эффективность разделения компонентов. Основными задачами твердого носителя неподвижной жидкой фазы являются:
В принципе носителем неподвижной жидкой фазы может быть любой тонко измельченный твердый материал, стабильный и инертный при рабочей температуре колонки. Однако достигаемая эффективность разделения зависит также и от следующих трех факторов:
Влияние степени однородности заполнения колонки насадкой на эффективность разделения заключается в следующем. Из уравнения Ван-Деемтера следует, что, для того чтобы число теоретических тарелок в расчете на метр колонки было достаточно большим, следует создавать условия для снижения величины коэффициента А. В этой связи следует стремиться, по возможности, к равномерному заполнению колонки частичками насадки. При этом использовать геометрически правильные частицы носителя, например, частицы сферической формы, так как в такой упорядоченной насадке путь газа-носителя оказывается менее извилистым, чем в нерегулярной сформированной насадке. Таким образом, хроматографирование следует проводить на носителе с достаточно узким распределением частиц по размерам. Используя такой носитель неподвижной жидкой фазы, можно получить колонки с воспроизводимым временем удерживания и одинаковыми характеристиками разделения. Установлено, что обеспечить равномерную упаковку колонки при малом сопротивлении потока газа-носителя и малом времени удерживания можно, используя фракции твердого носителя с диаметром частиц в интервале 0.005 – 0.8 мм, причем оптимальной является фракция 0.12 – 0.30 мм. Для повышения эффективности разделения целесообразно использовать более узкие фракции: 0.120.15, 0.150.18, 0.180.25 и 0.250.30 мм. При этом отмечено, что наиболее короткое время удерживания, откорректированное с учетом градиента давления, наблюдается в том случае, если отношение диаметра колонки к размеру частицы твердого носителя близко к 25. Для колонок диаметром 6 мм это соответствует зернам размером 0.24 мм, а для колонок диаметром 4 мм – зернам размером 0.16 мм. Хорошие результаты были получены при разделении на аналитических колонках, заполненных носителем с зернами размером по крайней мере 1/8 диаметра колонки. Поэтому колонки с внутренним диаметром до 2.5 мм и длиной до 2 м заполняют, как правило, частицами размером от 0.12 до 0.16 мм, более длинные колонки – носителем с частицами размером 0.160.18 мм. Колонки большего диаметра следует заполнять носителем с размерами частиц 0.160.18 мм, если их длина составляет менее 2 м, и частицами диаметром 0.180.25 мм, если длина колонки превышает 2 м. Поскольку пленка неподвижной жидкой фазы должна иметь достаточно большую поверхность и по возможности равномерно распределяться на носителе, большое внимание следует уделять вопросам геометрической структуры поверхности носителя. Установлено, что допустимый нижний предел величины удельной поверхности носителя составляет около 0.5 м2/г. На носителях с большей удельной поверхностью (до 2 м2/г) разделяемые соединения часто адсорбируются, что приводит к асимметричным пикам. Однако следует заметить, что при применении носителей с большой удельной поверхностью, на коротких колонках также можно получить малое время удерживания и высокий коэффициент разделения, если разделять неполярные и малополярные вещества на полярных неподвижных фазах. Действительно, исправленное время удерживания пропорционально произведению величины удельной поверхности носителя на длину колонки: t 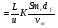 , (105) , (105) где L длина колонки; S удельная поверхность носителя; K коэффициент распределения вещества между фазами; u линейная скорость потока газа-носителя; mr масса носителя в колонке; vm объем подвижной фазы в колонке; df толщина пленки неподвижной жидкой фазы. Из приведенного уравнения видно, что можно получить приблизительно одинаковые значения времени удерживания и, следовательно, идентичные результаты разделения, если потерю эффективности разделения, обусловленную уменьшением длины колонки, компенсировать увеличением удельной поверхности носителя так, чтобы произведение LS оставалось постоянным. Еще одной важной характеристикой носителя, влияющей на эффективность разделения, являются размеры пор. Наилучшие параметры разделения имеют носители, у которых большая часть пор имеет диаметр от 0.510-3 до 1.510-3 мм. Дело в том, что при пропитке твердого носителя большая часть неподвижной жидкой фазы поглощается тонкой пористой структурой носителя, а остальную поверхность носителя покрывает тонкая пленка жидкости. При этом носитель выглядит сухим, но эффективность разделения оказывается хорошей. Эффективность разделения значительно ухудшается, если диаметр большинства пор превышает 1.510-3 мм или если вносится слишком большое количество неподвижной жидкой фазы, которая в этом случае объемно заполняет большие поры, образуя так называемые “масляные лужи”. Эти большие “масляные лужи” вследствие своей глубины имеют меньшее численное значение отношения величины поверхности к величине объема, чем более узкие поры, и поэтому растворенное анализируемое вещество из-за диффузии дольше находится в жидкости. В результате этого пики на хроматограммах уширяются, а эффективность разделения падает. Таким образом, поверхность носителя с преимущественно широкими порами плохо пропитывается неподвижной жидкой фазой. С другой стороны, слишком тонкопористый носитель, например, силикагель мало пригоден в качестве носителя, так как для слишком длинных и узких пор (диаметр от 0.2510-5 до 1.010-5 мм), заполненных неподвижной жидкой фазой, также характерно неблагоприятное отношение величины поверхности к объему (очень большое), что замедляет массообмен и ухудшает эффективность разделения. Адсорбционная активность твердого носителя неподвижной жидкой фазы отрицательно сказывается на разделении. Идеальным является такой твердый носитель, который адсорбционно инертен как по отношению к неподвижной жидкой фазе, так и к разделяемым компонентам. Отчасти этим требованиям отвечают только полиэтилен и политетрафторэтилен. Силикатные носители способны отщеплять воду от спиртов или приводить к перегруппировке реакционно способных производных пинена. Наряду с химическим взаимодействием твердого носителя с разделяемыми соединениями возможно и их физико-химическое взаимодействие, которое может быть обусловлено относительно большой внутренней поверхностью, наличием посторонних атомов и дефектами решетки. В таких случаях разделяемые соединения не только растворяются в неподвижной жидкой фазе, но и адсорбируются на поверхности носителя. Однако зачастую соответствующая изотерма адсорбции в отличие от изотермы растворения нелинейна, что приводит к образованию хвостов на хроматограмме. Это явление обнаруживается в том случае, если удельная поверхность носителя превышает величину 2 м2/г и если разделение поляр-ных соединений проводится на неполярных или малополярных жидких фазах. Причина этого явления состоит в том, что при наличии в смеси двух соединений различной полярности с активной поверхности твердого носителя может происходить вытеснение менее полярного (например, неподвижной жидкой фазы) соединения даже соединениями с меньшей молекулярной массой, если последние обладают достаточной полярностью. Следовательно, на адсорбционно активных твердых носителях вследствие нелинейности изотерм адсорбции время удерживания для системы анализируемое соединение – неполярная неподвижная жидкая фаза зависит от массы пробы; кроме того, остаточная адсорбционная активность тем интенсивнее влияет на процесс разделения, чем тоньше слой неподвижной жидкости, пропитывающей носитель, причем остаточная адсорбционная активность на разделение углеводородов влияет слабее, чем на разделение водных смесей, полиаминов, гликолей, спиртов, кислот, аминов, фенолов, кетонов, альдегидов, эфиров. На практике для того, чтобы устранить или снизить нежелательную адсорбционную активность твердого носителя неподвижной жидкой фазы, применяют один из нижеперечисленных способов химического или физического модифицирования поверхности. Химическое модифицирование твердого носителя проводится следующим образом:
Физическое модифицирование твердого носителя осуществляется следующим образом:
Таким образом, основные требования к носителям неподвижной жидкой фазы, применяемым в газо-жидкостной хроматографии, можно сформулировать следующим образом:
В качестве твердых носителей неподвижных жидких фаз предложено большое число материалов, однако, лишь немногие из них достаточно полно удовлетворяют указанным требованиям. Наибольшее распространение в практике получили следующие твердые носители: диатомовые носители, стеклянные микрошарики, силикагель, оксид алюминия, политетрафторэтилен, полимерные пористые носители. 13.2. Классификация основных носителей неподвижных жидких фаз Диатомовые носители Исходным материалом для носителей служит светло-серое или красновато-коричневое аморфное вещество, представляющее собой обломки панцирей микроскопических диатомовых водорослей – диатомит. Диатомовая горная порода состоит в основном из аморфного кремнезема, содержащего от 20 до 80 % физически связанной воды. Диатомит промывают водой, чтобы отделить загрязняющий материал, например песок, сушат и перемалывают. Прокаливанием во вращающейся печи, иногда в присутствии гидроксидов, удаляют органические соединения. Цвет диатомитов при этом изменяется, а его удельная поверхность уменьшается от 1240 (сырой продукт) до 15 м2/г. Далее диатомит измельчают, просеивают и, если необходимо, освобождают от оксидов железа. Из такого порошкообразного материала и готовят носитель для хроматографических колонок. Следует различать диатомовые носители первого и второго типа. Диатомовые носители первого типа характеризуются достаточно большой механической прочностью, величиной удельной поверхности порядка 4 м2/г, большой насыпной массой (г/см3). Вследствие пропорциональной зависимости между удельной поверхностью и адсорбционными свойствами носителя этот недоста-ток проявляется ярче, чем у носителей второго типа. В связи с этим носители первого типа применяются преимущественно для анализа смесей углеводородов. Представителями твердых носителей этого типа являются: стерхамол, рисорб, диатопорт, анакром, сферохром2, сферохром3, динохромN, ИНЗ600. Диатомовые носители второго типа характеризуются величиной удельной поверхности порядка 1 м2/г, малой насыпной массой и, как следствие, малой адсорбционной активностью. Наиболее часто в практике встречаются следующие представители этого типа носителей: кизельгур, хромосорб, целит, газохром, сферохром1, порохром, динохром, хроматон, инертон. Модифицирование носителей. Обработка кислотами. Установлено, что неорганические загрязнения кислотами отмываются полнее, чем гидроксидами. Предварительную обработку кислотами рекомендуется проводить перед силанизацией, так как в этом случае носитель становится более инертным, чем в результате одной лишь силанизации. Обработка кислотами (чаще всего соляной) проводится следующим образом: 100 г твердого носителя смешивают с 500 мл дистиллированной воды в стакане емкостью 1 л. Через 2 минуты взвесь мелких частиц декантируют. Процесс повторяют до удаления практически всех мелких пылевидных частиц. Носитель отфильтровывают на воронке Бюхнера, отсасывают досуха и переносят в стакан емкостью 500 мл. Добавляют 250 мл концентрированной соляной кислоты, тщательно перемешивают, дают 2 минуты отстояться, декантируют и снова добавляют 150 мл кислоты. Смесь перемешивают и оставляют стоять на ночь. Затем смесь тщательно взмучивают и декантируют. Декантация проводится четырежды, добавляя каждый раз 300 мл воды. Далее носитель отфильтровывают на воронке Бюхнера, промывают водой до нейтральной реакции, отсасывают досуха и сушат при 150 оС на воздухе в течение суток. Обработка гидроксидами. Обработку гидроксидами диатомовых носителей следует проводить при разделении основных соединений аминов, диаминов, пиридинов, хинолина, гуанидина, метиламина, эпоксипроизводных. Отмечено, что обработка гидроксидами разрушает каталитические центры носителя и снижает их активность. На практике твердый носитель тщательно пропитывают метанолом и полученную массу перемешивают с 6-процентным метанольным раствором гидроксида в течение часа в роторном испарителе без нагревания. Затем отгоняют в вакууме растворитель, и высушенный продукт просеивают. При использовании обработанных гидроксидами носителей в практике следует учитывать способность свободных гидроксидов разлагать или адсорбировать многие из разделяемых соединений. Обработка силанизирующими реагентами. Свободные группы  Si-OH, расположенные на поверхности носителя, оказывают мешающее действие, особенно в тех случаях, если на носитель нанесено очень небольшое количество неподвижной жидкой фазы или если жидкая фаза почти неполярна, а разделяемые соединения характеризуются значительной полярностью. Si-OH, расположенные на поверхности носителя, оказывают мешающее действие, особенно в тех случаях, если на носитель нанесено очень небольшое количество неподвижной жидкой фазы или если жидкая фаза почти неполярна, а разделяемые соединения характеризуются значительной полярностью.Остаточную активность носителя можно снизить еще в большей степени, если провести замещение атома водорода в группах Si-OH на органосилильные радикалы.Одним из вариантом модифицирования поверхности является обработка носителя диметилдихлорсиланом или триметилхлорсиланом: SiOH + (CH3)3SiCl SiOSi(CH3)3 + HCl.Силанизация гексаметилдисилазаном протекает по схеме: SiOH + (CH3)3SiNHSi(CH3)3 SiOSi(CH3)3 + (CH3)3SiNH2SiOH + (CH3)3SiNH2 SiOSi(CH3)3 + NH3(CH3)3SiNHSi(CH3)3 + H2O (CH3)3SiOSi(CH3)3 + NH3. Последнее уравнение уравнение медленно протекающей побочной реакции. В результате проведения силанизации удельная поверхность носителя может несколько снизиться, однако если сам исходный диатомит малоактивен, то силанизация позволяет получить носитель, пригодный для эффективного разделения большинства смесей. У таких носителей имеется лишь один недостаток: так как покрытая кремнийорганическим соединениями поверхность носителя оказывается гидрофобной, она слабее удерживает такие полярные неподвижные жидкие фазы, как глицерин или диглицерин, в результате чего на таких неподвижных жидких фазах, прежде всего при повышенных загрузках колонок, эффективность разделения невелика. Отмытые кислотой, гидроксидами и силанизированные носители специальным образом маркируются. Например, носитель газохром-Q ABW-DMCS промыт кислотой, гидроксидом, водой и обработан диметилхлорсиланом. Стеклянные микрошарики При использовании стеклянных микрошариков в качестве носителей неподвижной жидкой фазы следует учитывать, что они обладают определенными адсорбционными свойствами. Их адсорбционную активность можно снизить путем использования стекла, не содержащего кальция, и блокированием групп SiOH диметилдихлорсиланом, как и в случае диатомовых носителей.Вследствие малой удельной поверхности стеклянных шариков на них возможно нанесение только очень небольшого количества неподвижной жидкой фазы. Максимально возможное количество жидкости, удерживающейся на поверхности шарика, зависит от его радиуса, величин поверхностного натяжения и плотности жидкости и составляет от 0.05 до 3 %. При этом следует учитывать, что вследствие большой плотности стеклянного носителя (около 2 г/см3) содержание 1 % неподвижной фазы на шариках соответствует 10 % содержанию на диатомовом носителе хромосорбеW (плотность 0.22 г/cм3). При тщательной предварительной обработке стеклянного носителя и низкой концентрации неподвижной жидкой фазы (до 0.02 %) минимальное значение высоты эквивалентной теоретической тарелке может составить 0.5 мм, при этом наблюдается минимальная адсорбционная активность. Силикагель Необработанный силикагель, изготовленный из кремниевой кислоты, имеет очень большую поверхность и тонкие поры, что приводит к проявлению высокой адсорбционной активности и не дает возможности применять его в качестве носителя неподвижной жидкой фазы. Тем не менее вследствие постоянства своего химического состава, силикагель в большей степени пригоден в качестве носителя, независимого от загрузки пробой, чем, например, диатомит, содержащий целый ряд загрязнений (примеси железа, алюминия, магния, кальция), если можно уменьшить величину поверхности силикагеля, расширить слишком узкие поры, добиться равномерного распределения пор по диаметрам и дезактивировать группы SiOH.Носитель с хорошими свойствами получается путем обработки силикагеля водой в автоклаве с последующим замещением ОНгрупп на радикалы ОSi(CH3)3. Оксид алюминия Оксид алюминия, как и необработанный силикагель, проявляет большую адсорбционную активность и взаимодействует с разделяемыми веществами, поэтому его селективность в значительной мере зависит от степени пропитки неподвижной жидкой фазой. Политетрафторэтилен Политетрафторэтилен является важнейшим носителем из числа органических носителей. Он превосходит другие материалы по термостойкости, позволяет работать при температурах до 200 оС. Выше этой температуры форма частиц носителя меняется, что приводит к снижению эффективности разделения. Начиная с температуры 290 оС, политетрафторэтилен разлагается с выделением ядовитого перфторизобутена. Поэтому перегревания носителя до такой температуры допускать нельзя. Основным преимуществом политетрафторэтилена является чрезвычайно низкая химическая активность: он реагирует только с расплавленными щелочными металлами и элементным фтором и не проявляет никакой каталитической и адсорбционной активности. Другие носители Для разделения сильнополярных низкомолекулярных соединений часто используют пористые носители на основе сополимеров стирола и дивинилбензола, известных под названиями полисорб 1,2. 13.3. Неподвижные жидкие фазы Неподвижная жидкая фаза является ответственной за разделение смесей газов или паров на отдельные компоненты, которое должно осуществляться достаточно быстро и достаточно эффективно. В этой связи вещества, используемые в качестве неподвижных жидких фаз, должны отвечать определенным требованиям по следующим параметрам:
Химическая активность Неподвижная жидкая фаза не должна вступать в необратимые реакции ни с газом-носителем, ни с компонентами разделяемой смеси, ни с носителем неподвижной жидкой фазы. Например, при применении в качестве газа-носителя воздуха или иного содержащего кислород газа возможно окисление чувствительных к воздействию кислорода неподвижных жидких фаз, часто с образованием соединений, обладающих меньшей разделительной способностью. В таком случае либо заменяют неподвижную жидкую фазу на более устойчивую к окислению, либо применяют газ-носитель, свободный от кислорода. Если, например, используется носитель, характеризующийся достаточной адсорбционной активностью, то ее следует снизить настолько, чтобы состав неподвижной жидкой фазы не изменялся. Химические реакции, проходящие с отдельными компонентами разделяемой смеси, изменяют ее состав, и на хроматограмме появляются пики соединений, отсутствующих в исходной смеси или, наоборот, отсутствуют пики, заведомо содержащихся соединений, которые необратимо удерживаются неподвижной жидкой фазой. Следует, однако, иметь в виду, что иногда желательно образование лабильных соединений присоединения, так как они обеспечивают исключительно высокую селективность. Давление паров и термостойкость В качестве неподвижных жидких фаз можно в принципе использовать любые органические соединения, однако на практике чаще всего используются термически стабильные в условиях работы хроматографической колонки вещества. Термостабильность неподвижной жидкой фазы определяется двумя факторами:
Поскольку в газо-жидкостной хроматографии разделение компонентов смеси производится только в таком температурном интервале, когда нанесенная на твердый носитель неподвижная жидкая фаза представляет собой жидкость, каждая из используемых жидких фаз должна быть охарактеризована величиной нижнего и верхнего температурного предела ее использования. Нижний температурный предел использования неподвижной жидкой фазы характеризуется величиной температуры плавления вещества, используемого в качестве неподвижной жидкой фазы. Однако известно, что температура плавления вещества зависит от степени его чистоты. В этой связи при нанесении на твердый носитель небольшого количества неподвижной жидкой фазы температура плавления этого вещества может повышаться на несколько градусов вследствие адсорбционного взаимодействия неподвижной жидкой фазы с поверхностью носителя. Поэтому в качестве нижнего температурного предела использования рекомендуется выбирать температуру на 510 оС выше температуры плавления чистого вещества. Следует учитывать, что некоторые вещества, используемые как неподвижные жидкие фазы, не могут быть охарактеризованы определенной величиной нижнего температурного предела: к ним относятся вещества, для которых характерно стекловидное состояние, полимеры с большой молекулярной массой. Температура плавления вещества зависит от плотности упаковки его молекул: вещества с относительно регулярной, плотной упаковкой характеризуются более высокой температурой плавления. В этой связи для снижения нижнего температурного предела целесообразно использовать соединения, углеродные цепочки молекул которых содержат большое число разветвлений. Наличие в молекуле атомных групп, вступающих в электростатическое или специфическое взаимодействие, повышает температуру плавления вещества. Особенно высокие значения температур плавления наблюдаются для соединений, молекулы которых содержат большое число гидроксильных групп. Верхний температурный предел использования неподвижной жидкой фазы зависит от факторов, связанных с потерей неподвижной жидкой фазы и с чувствительностью детектора. Потери неподвижной жидкой фазы из колонки могут быть обусловлены двумя процессами:
Потери за счет испарения свойственны в основном мономерным неподвижным фазам, а потери за счет термодеструкции наблюдаются чаще всего для полимерных неподвижных жидких фаз. Верхний температурный предел предложено определять такой температурой, при которой в течение трех месяцев непрерывного использования потеря неподвижной жидкой фазы по массе составляет около 2 %. Если нанести на носитель неподвижную жидкую фазу в количестве на 10 % больше оптимального, то испарение из колонки 1520 % неподвижной жидкой фазы практически не изменит ее эффективности, и такие колонки могут служить до трех лет, что вполне допустимо для аналитических целей. Верхний температурный предел зависит также от условий разделения. Так, например, оптимальное количество неподвижной жидкой фазы на белом диатомовом носителе составляет 1015 %. Если же для разделения необходимо применять колонки с небольшим количеством неподвижной жидкой фазы на носителе, потери 23 % неподвижной фазы изменяют качество разделения. Поэтому для подобных колонок верхний температурный предел по сравнению с основными данными снижается на 3050 оС. Верхний температурный предел полимерных неподвижных жидких фаз определяется температурой разрыва наименее стойких связей в молекуле полимера, при этом образуются низкомолекулярные летучие соединения, уносимые газом-носителем из колонки. Для неподвижных фаз полимерной природы минимальная температура разрыва наблюдается для эфирных связей. Обычная эфирная связь начинает разрушаться уже при температуре 200 оС; эта температура и является предельной для использования многочисленных полиэфирных неподвижных жидких фаз. Большей стабильностью обладают полимеры, содержащие простые связи углеродуглерод. Такие фазы можно использовать до 350 оС, однако существенным недостатком этих полимеров является их высокая температура плавления или размягчения. Наиболее термостабильными неподвижными жидкими фазами являются кремнийорганические полимеры, например, метилсиликоны (верхний температурный предел 300350 оС) и карборансилоксановые полимеры, основой полимерной цепи которых служит сферическая ячейка, в которую входят атомы бора, кремния и углерода (верхний температурный предел 400 оС). Практика использования неподвижных жидких фаз для длительной аналитической работы показывает, что необходимо работать с неподвижными фазами при температурах на 2030 оС ниже основных верхних температурных пределов. И, наконец, на практический верхний температурный предел влияет также и чувствительность используемого детектора: чем выше чувствительность детектора, тем ниже верхний температурный предел использования данной неподвижной жидкой фазы. Так, в случае пламенно-ионизационных детекторов на их максимальной чувствительности реальный верхний температурный предел использования оказывается на 5060 оС ниже основного значения. Размеры молекул Влияние размеров молекул неподвижной жидкой фазы следует особенно учитывать при использовании полимерных материалов, состоящих из большого числа соединений с различной молекулярной массой. Такие полимеры в зависимости от состава исходной полимеризационной смеси могут иметь различные свойства: различное давление паров, температуру кипения и состав продуктов разложения. Поэтому важно, чтобы распределение по молекулярным массам было достаточно узким, что исключает вероятность испарения молекул с меньшей молекулярной массой, которое приводит к снижению количества неподвижной жидкой фазы и изменению ее свойств. Вязкость Если разделение проводится на неподвижных жидких фазах с очень высокой вязкостью, хорошие результаты можно получить только при повышении температуры и соответствующем снижении вязкости, так как сопротивление переносу вещества пропорционально вязкости неподвижной жидкой фазы. Исключение составляют неполярные линейные полимеры, для которых увеличение длины цепи мало влияет на диффузию маленьких молекул. Способность к образованию пленок Хорошая эффективность разделения достигается в случае, если поверхность носителя покрыта равномерной пленкой неподвижной жидкой фазы. Образование такой пленки возможно лишь при такой температуре, при которой неподвижная жидкая фаза находится в жидком состоянии и вязкость ее не слишком велика, кроме того, химическая структура молекул неподвижной жидкой фазы должна обеспечивать минимальный угол смачивания на поверхности раздела носитель – неподвижная жидкая фаза. Это возможно в том случае, если обе фазы имеют близкое химическое строение. Поэтому следует предпочтительно использовать силанизированные носители, т.е. такие носители, поверхность которых частично покрыта алкилсилильными группами. На такой поверхности легче образуются пленки многих органических соединений (исключение составляют соединения, в молекулах которых содержатся группы ОН или NH2). Способность к растворению разделяемых соединений Для того, чтобы на жидкой фазе могло происходить разделение смесей, она должна растворять их с тем, чтобы они перемещались вдоль колонки не слишком быстро. Поэтому неподвижные жидкие фазы близкие по своей химической природе к разделяемым соединениям, применяются преимущественно для исследования смесей, компоненты которых относятся к одному и тому же гомологическому ряду, так как для них характерны различия в давлении паров и результаты хроматографирования зависят исключительно от эффективности разделения. Разделительные свойства Если требуется разделить соединения, имеющие одинаковую темтепературу кипения, но принадлежащие к различным гомологическим рядам, следует пользоваться неподвижной жидкой фазой, характеризующейся достаточной селективностью. Неподвижная жидкая фаза обладает селективностью, если в процессе разделения веществ, принадлежащих к различным гомологическим рядам с одинаковой температурой кипения, обнаруживается существенное различие в их относительном удерживании. Разделение происходит, например, в том случае, если один из компонентов смеси хорошо растворяется в неподвижной жидкой фазе, а другой – плохо. При разделении соединений одного и того же класса следует отказываться от правила подобия. Здесь наибольшим разделительным действием обладают неподвижные жидкие фазы, отличающиеся от разделяемых соединений полярностью. Например, углеводороды с одинаковой температурой кипения интенсивнее разделяются на таких полярных неподвижных фазах, как нитрилы, а разделение полярных соединений успешно проходит на малополярных силиконовых маслах или неполярных фазах на основе углеводородов (парафиновое масло, сквалан). Особенно высокую селективность проявляют неподвижные жидкие фазы, образующие с отдельными компонентами анализируемой смеси слабосвязанные аддукты, как, например, нитрат серебра, удерживающий олефины, или пикраты, селективно присоединяющие ароматические соединения. 13.4. КЛАССИФИКАЦИЯ НЕПОДВИЖНЫХ ЖИДКИХ ФАЗ В газо-жидкостной хроматографии априорный выбор оптимальной неподвижной жидкой фазы для решения задачи разделения конкретной смеси – довольно сложная задача. Это объясняется тем, что теория растворов к настоящему времени еще не разработана до такой степени, чтобы можно было охарактеризовать все возможные типы взаимодействий разделяемых веществ с неподвижной жидкой фазой легко определяемыми константами или коэффициентами. Достаточно обоснованный выбор оптимальной неподвижной жидкой фазы может быть осуществлен только лишь при наличии рациональной классификации этих фаз. Классификация неподвижных жидких фаз по величинам относительной полярности Исторически первой была классификация неподвижных жидких фаз, основанная на величинах их относительной полярности, предложенная Роршнейдером. Относительная полярность неподвижной жидкой фазы определяется из соотношения исправленных удерживаемых объемов вещества, выступающего в качестве стандарта полярного растворенного вещест-ва, и вещества, выступающего в качестве стандарта неполярного растворенного вещества, измеренных на колонках с полярной исследуемой неподвижной жидкой фазой и стандартной неподвижной жидкой фазой, полярность которой принята за нуль: 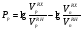 , (106) , (106)где Рр полярность полярной неподвижной жидкой фазы р;VpRX, VpRH удерживаемые объемы компонентов RX и RH на полярной неподвижной жидкой фазе соответственно; VnRX, VnRH удерживаемые объемы компонентов RX и RH на стандартной неполярной неподвижной жидкой фазе соответственно. Для любого другого анализируемого вещества, не являющегося стандартным, соотношение удерживаемых объемов на неподвижной жидкой фазе р можно представить в виде: 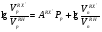 , (107) , (107)где АRX’ характеристическая константа растворенного вещества RX’. Уравнение (106) позволяет составить шкалу полярности исследуемых неподвижных жидких фаз. Для этого выбирают стандартные вещества – полярное (RX) и неполярное (RH) и определяют их удерживаемые объемы на неполярной и исследуемой полярной неподвижных жидких фазах. Роршнейдер предложил применять в качестве стандартных веществ бутадиен (полярное) и бутан (неполярное). В качестве неподвижной жидкой фазы с величиной относительной полярности равной нулю был предложен сквалан (разветвленный насыщенный углеводород с числом атомов углерода в молекуле, равным 30). Наиболее полярной неподвижной жидкой фазой, которой Роршнейдер установил полярность, равную 100 единицам, выбран , ’ – оксидипропионитрил. Тогда, приняв в уравнении (107) величину относительной полярности Рр, равную 100 для ,’-оксидипропионитрила, можно рассчитать величину характеристической константы АRX^ для соединений олефинового ряда из уравнения 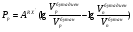 . (108) . (108)Можно использовать и графический способ определения величины относительной полярности исследуемой неподвижной жидкой фазы. В табл. 12 приведена шкала относительной полярности некоторых неподвижных жидких фаз, рассчитанная по удерживаемым объемам бутадиена и н-бутана при 30 оС в случае разделения молекул олефинового ряда. Т а б л и ц а 12 Шкала относительной полярности неподвижных жидких фаз
|