бизнес план. Аяулым тема. Изучение молекулярных предикторов сосудистого риска опосредованного эндотелиальной дисфункцией у людей с сахарным диабетом 2 типа
 Скачать 1.19 Mb. Скачать 1.19 Mb.
|

ИНСУЛИНОВАЯ РЕЗИСТЕНТНОСТЬ И ГОРМОНЫ ИНКРЕТИНОВОГО РЯДАНарушение секреции инкретиновых гормонов и сниженный инкретиновый эффект являются причинами прогрессирования гипергликемии у пациентов с классическим СД2. Стимуляция выработки гормонов инкретинового ряда, в частности глюкагоноподобного пептида-1 (ГПП-1), ведет к компенсации углеводного обмена [54, 55]. Воздействие на этот механизм реализуется при назначении инкретин-направленной терапии [56, 57]. Для того чтобы проверить гипотезу о возможной роли гормонов инкретинового ряда в ремиссии СД2, было проведено клиническое исследование, в которое были включены пациенты с длительным анамнезом СД2 и ожирения (более 10–15 лет), получавшие терапию агонистом рецептора ГПП-1 лираглутидом в дозе, имитирующей максимальный инкретиновый эффект. Через 3–4 мес после получения препарата у пациентов было отмечено среднее снижение массы тела на 6,2 кг, гликированного гемоглобина крови (HbA1c) на 1,1% и повышение М-индекса с 1,74 мг/кг/мин до 2,52 мг/кг/мин, что говорит о снижении выраженности ИР и перспективах использования инкретин-направленной терапии для коррекции ИР у больных с СД2. ЗАКЛЮЧЕНИЕОбщая последовательность развития событий в патогенезе СД2 довольно понятна (рис. 2). Первичные факторы риска, включающие ожирение, воспаление и стресс различной природы, ведут к развитию ИР в клетках-мишенях инсулина. В классическом варианте в адипоцитах жировой ткани все факторы риска ИР действуют через единый механизм, связанный с латентным воспалением и хронической активацией стресс-зависимых киназ, таких как JNK и IKK. Они фосфорилируют субстрат инсулинового рецептора IRS, нарушая активацию инсулинового каскада и выход глюкозного транспортера Glut4 на поверхность клеток (см. рис. 1В). На молекулярном уровне ИР проявляется в снижении инсулинзависимого фосфорилирования компонентов инсулинового каскада, киназы Akt и белка AS160 (см. рис. 1Г). Наши данные подтверждают, что сайт-специфичное фосфорилирование Akt, AS160 и JNK может служить молекулярным маркером ИР в адипоцитах. 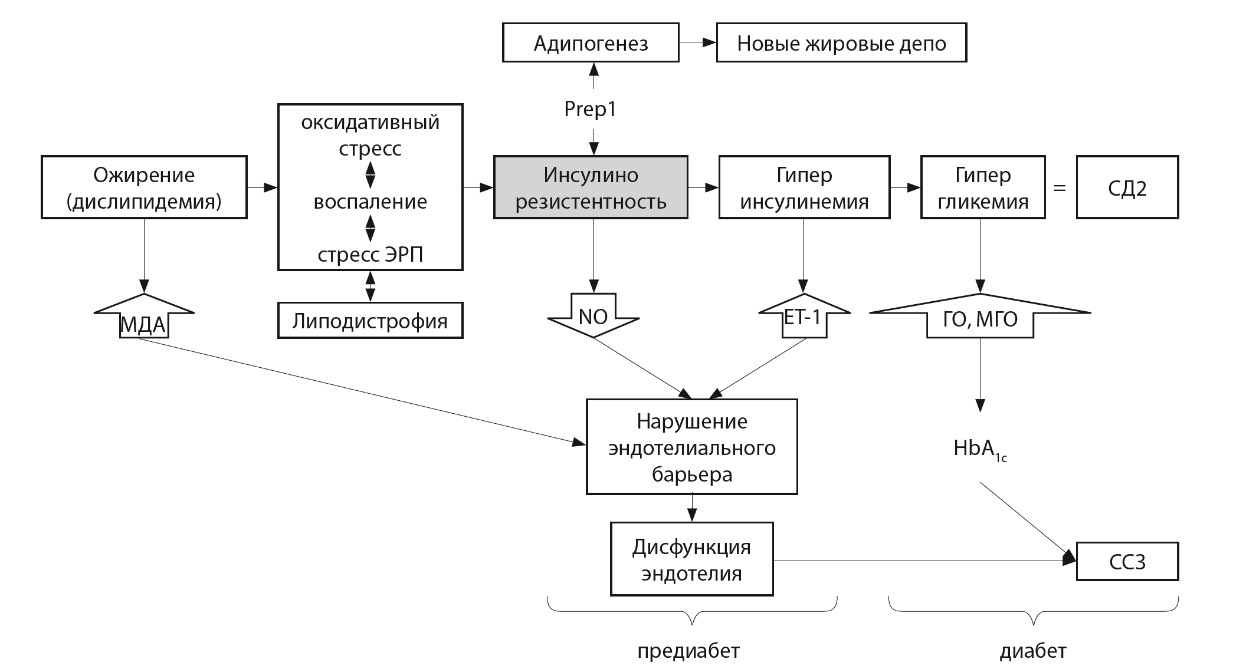 Рис. 2. Общая схема патогенеза сахарного диабета 2 типа и ассоциированных сердечно-сосудистых осложнений. Липодистрофия представляет неклассический вариант патогенеза СД2, по крайней мере, в отношении жировой ткани. Наши и другие исследования определяют четкую связь между липодистрофией и ИР, но причинно-следственные взаимоотношения и патогенез СД2 при липодистрофии остаются малопонятными и требуют отдельных исследований. Избыток свободных жирных кислот вследствие ожирения является важной причиной ИР и карбонильного стресса, повышая риск дисфункции сосудистого эндотелия и сердечно-сосудистых осложнений уже на первых стадиях патогенеза СД2. Активируя толл-подобные рецепторы TLR4, СЖК запускают воспалительные каскады и развитие ИР в адипоцитах. Кроме того, в клетках СЖК подвергаются перекисному окислению с образованием МДА как одного из продуктов. Физически воздействуя на эндотелий и модифицируя ряд белков, МДА изменяет структуру цитоскелета и межклеточных контактов, нарушая барьерные свойства эндотелия и повышая риск развития отеков, гипертонических осложнений и ангиопатий. Прогрессирование патологического процесса ведет к устойчивой гипергликемии и карбонильному стрессу за счет активных продуктов распада глюкозы. ГО и МГО химически модифицируют другие белки плазмы и клеток крови, что согласуется с повышением HbA1c. sHSP защищают клетки от окислительного стресса и апоптоза на первых этапах патогенеза СД2, препятствуя агрегации денатурированных белков и развитию стресса ЭПР. Вместе с тем sHSP являются мишенью карбонильного стресса при гипергликемии, что ведет к изменению их структуры и функциональных свойств, уменьшению шапероноподобной активности и защитного действия. Тем самым sHSP могут опосредовать обратную связь от конечных к начальным этапам патогенеза СД2, усиливая эффекты оксидативного стресса и стресса ЭПР. Персистирующая ИР запускает отложенные, длительные адаптационные процессы в организме; они ведут к перестройке всего метаболизма в процессе развития СД2. Такие длительные изменения закрепляются на транскрипционном уровне, где ключевую роль играют различные транскрипционные факторы. При этом направление перестроек часто определяется совокупным воздействием регуляторов транскрипции и внешних факторов. Например, в условиях гипергликемии может усиливаться экспрессия Prep1 в миоцитах, что снижает чувствительность мышц к инсулину и утилизацию ими глюкозы, но выполняет защитную функцию. Аналогичную роль может играть Prep1 в адипоцитах, где он также ослабляет чувствительность к инсулину и захват глюкозы, предотвращая жировую перегрузку адипоцитов. В этой связи важно, что Prep1 участвует в регуляции адипогенеза, то есть он также подавляет процесс формирования новых жировых депо. В совокупности системный эффект Prep1 можно расценивать как компенсаторное отключение инсулиновой зависимости клеток в условиях гиперинсулинемии и пищевой перегрузки. Дальнейшие исследования должны быть направлены на валидацию Prep1 как терапевтической биомишени в животных моделях и поиск способов направленного воздействия на его экспрессию в инсулинчувствительных тканях. Очевидно, что патогенез СД2 гораздо более сложен, чем схематично показанный на рис. 2. Определение других его участников и механизмов, безусловно, требует продолжения исследований. |