Каталитическая функция. Большинство известных в настоящее время ферментов, называемых биологическими катализаторами, является белками. Транспортная функция
 Скачать 1.95 Mb. Скачать 1.95 Mb.
|

|
Трансаминирование — биохимическая ферментативная реакция обратимого переноса аминогруппы с аминокислоты на кетокислоту без промежуточного образования аммиака. Ферменты, катализирующие процесс, назвали трансаминазами, или аминотрансферазами. Продуктами чаще всего являются аланин, аспарагин и глутамат, так как соответствующие им кетокислоты образуются в процессе метаболизма углеводов. Трансаминирование играет важную роль в процессах мочевинообразования, глюконеогенеза, путях образования новых аминокислот. Трансаминирование аминокислот с образованием глутаминовой кислоты в сочетании с ёё дезаминированием НАД(Ф)-зависимой глутаматдегидрогеназой называется непрямым дезаминированием аминокислот (трансдезаминирование). 82. Обмен безазотистого остатка аминокислот. Гликогенные и кетогенные аминокислоты. Синтез глюкозы из аминокислот (глюкозо-аланиновый цикл). Синтез аминокислот из продуктов обмена глюкозы: синтез серина, глицина. Роль фолиевой кислоты в обмене этих аминокислот. Использование глицина для синтеза различных веществ в организме. За сутки у человека распадаются примерно 100г АК. Катаболизм всех АК сводится к образованию шести веществ, вступающих в общий путь катаболизма: ПВК, ацетил-КоА, α-кетоглутарат, сукцинил-КоА, фумарат и ЩУК. Эти вещества окисляются в ЦТК для образования АТФ или используются для синтеза глюкозы и кетоновых тел. Гликогенные аминокислоты -АК, которые превращаются в ПВК и промежуточные продукты ЦТК (а-КГ, сукцинил-КоА, фумарат, ЩУК). Они через ЩУК, используются в глюконеогенезе (ала, асн, асп, гли, глу, глн, про, сер, цис, арг, гис, вал, мет, тре). Кетогенные аминокислоты – АК, которые в процессе катаболизма превращаются в ацетоацетат (Лиз, Лей) или ацетил-КоА (Лей) и могут использоваться в синтезе кетоновых тел. Смешанные (гликкетогенными) аминокислоты – АК, при катаболизме которых образуются метаболит цитратного цикла и ацетоацетат (Три, Фен, Тир) или ацетил-КоА (Иле). Эти АК используются для синтеза глюкозы и кетоновых тел. 83. Декарбоксилирование аминокислот. Биогенные амины: гистамин, серотонин, гаммааминомасляная кислота, катехоламины. Их происхождение, функции, расщепление. Образование токсических аминов в толстом кишечнике, их обезвреживание в печени. Процесс отщепления карбоксильной группы аминокислот в виде CO2 получил название декарбоксилирования. В животных тканях установлено декарбоксилирование следующих аминокислот и их производных: тирозина,триптофана, 5-окситриптофана, валина, серина, гистидина, глутаминовой и γ-оксиглутаминовой кислот, 3,4-диоксифенилаланина, цистеина, аргинина, орнитина, S-аденозилметионина и α-аминомалоновой кислоты. Помимо этого, у микроорганизмов и растений открыто декарбоксилирование ряда других аминокислот. Биогенные амины — вещества, обычно образующиеся в организме животных или растений из аминокислот при их декарбоксилировании (удалении карбоксильной группы) ферментами декарбоксилазами и обладающие высокой биологической активностью. К биогенным аминам относятся дофамин, норадреналин и адреналин (синтезируются изначально из аминокислоты тирозина), серотонин, мелатонин и триптамин и многие другие соединения. В организме животных многие биогенные амины выполняют роль гормонов и нейромедиаторов. Разлагаются в организме при участии ферментов аминоксидаз. Реакции декарбоксилирования являются необратимыми. Они катализируются специфическими ферментами – декарбоксилазами аминокислот, отличающимися от декарбоксилаз α-кетокислот как белковым компонентом, так и природой кофермента. 84. Обмен фенилаланина и тирозина. Образование катехоламинов, гормонов щитовидной железы, меланина. Нарушения процессов распада тирозина: фенилкетонурия, алкаптонурия, альбинизм. Фенилаланин – незаменимая аминокислота, так как в клетках животных не синтезируется ее бензольное кольцо. Метаболизм метионина осуществляется по 2-м путям: включается в белки или превращается в тирозин под действием специфической монооксигеназы – фенилаланингидроксилазы. Кроме использования в синтезе белков, тирозин в разных тканях выступает предшественником таких соединений как катехоламины, тироксин, меланин и др. В щитовидной железе из тирозина синтезируются гормоны тироксин и трийодтиронин. В мозговом веществе надпочечников и нервной ткани тирозин является предшественником катехоламинов. Промежуточным продуктом их синтеза является ДОФА. Однако в отличие от меланоцитов, гидроксилирование тирозина осуществляется под действием тирозингидроксилазы, которая является Fe2+- зависимым ферментом, и его активность регулирует скорость синтеза катехоламинов. 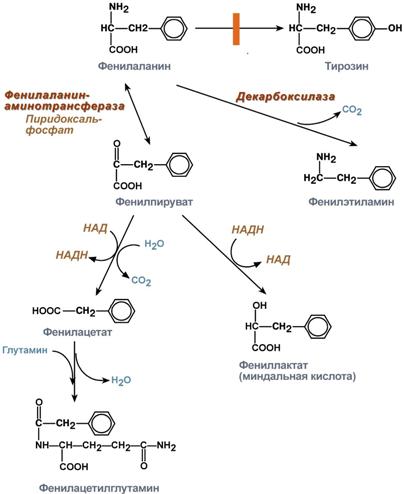 85. Лабильность дезаминирования в гепатоцитах ребенка. Повышенная концентрация аминокислот в крови и моче ребенка раннего возраста. Своеобразие обмена некоторых аминокислот. Фенилаланин, тирозин. О гипераминоацидуриях говорят в том случае, когда выведение одной или нескольких аминокислот с мочой превышает физиологические значения. В зависимости от происхождения можно выделить: 1. метаболические или преренальные и 2. ренальные аминоацидурий. При метаболических аминоацидуриях одной или нескольких аминокислот образуется больше, нежели в норме, или метаболизируется меньшее их количество. Фенилкетонурия. Фенилпировиноградная олигофрения (болезнь Фёллинга). Энзимопатия, наследуемая по аутосомно-рецессивному типу. Ее биохимической сущностью является невозможность превращения фенилаланина в тирозин вследствие отсутствия фермента фенилаланин-оксидазы. Клинические проявления этой аномалии связаны с выраженным повреждением мозга, сопровождающимся умственной отсталостью. Это нередкое заболевание - одна из наиболее частых причин олигофрении. Среди населения встречается с частотой 1:10 000-1: 20 000. Алкаптонурия. Заболевание характеризуется темно-коричневой окраской мочи, которая появляется при стоянии на воздухе. Наследственная энзимопатия, у больных отсутствует фермент гомогентизиназа. Гомогентизиновая кислота, выделяемая в большем количестве, на воздухе окисляется, приобретая коричневый цвет. Пеленки и нижнее белье ребенка также окрашиваются, что облегчает постановку диагноза. Кроме описанной выше особенности мочи, при этой аномалии имеются только два других симптома: появляющаяся в более позднем возрасте артропатия и синеватая окраска хрящей, легко обнаруживаемая на ушной раковине. Лечения нет. Альбинизм также является наследственной аномалией обмена ароматических аминокислот. При этом отсутствует энзим тирозиназа, который катализирует превращение тирозина в ДОФА - диоксифенилаланиц. Так как ДОФА - основа для синтеза меланина, то носители аномалии светлокожие, светловолосые люди, у которых через лишенную пигментации радужную оболочку просвечивает красноватая сосудистая сеть. Больным следует избегать прямого солнечного света. 86. Генетически детерминированные патологии аминокислот у детей. Нарушения обмена триптофана, тирозина, фенилаланина. Нарушения обмена фенилаланина. Фенилаланин в норме необратимо окисляется в тирозин. Если же в печени нарушается синтез необходимого для этого фермента фенилаланингидроксилазы, то окисление фенилаланина идет по пути образования фенилпировиноградной и фенилмолочной кислот — развивается фенилкетонурия. Однако этот путь обладает малой пропускной способностью и поэтому фенилаланин накапливается в большом количестве в крови, тканях и цереброспинальной жидкости, что впервые же месяцы жизни ведет к тяжелому поражению центральной нервной системы и неизлечимому слабоумию. Из-за недостаточного синтеза тирозина снижается образование меланина, что обусловливает посветление кожи и волос. Кроме того, при увеличенной выработке фенилпировиноградной кислоты тормозится активность фермента (дофамингидроксилазы), необходимого для образования катехоламинов (адреналина, норадреналина). Поэтому тяжесть наследственного заболевания определяется комплексом всех этих нарушений. Нарушения обмена тирозина. Обмен тирозина осуществляется несколькими путями. При недостаточном превращении образовавшейся из тирозина парагидроксифенилпировиноградной кислоты в гомогентизиновую первая, а также тирозин выделяются с мочой. Это нарушение носит название тирозиноза. Если же задержка окисления тирозина происходит в момент превращения гомогентизиновой кислоты в малеилацетоуксусную, развивается алкаптонурия. Фермент, окисляющий гомогентизиновую кислоту (оксидаза гомогентизиновой кислоты), образуется в печени. В норме он настолько быстро разрывает ее гидрохиноновое кольцо, что кислота "не успевает" появиться в крови, а если и появляется, то быстро выводится почками. При наследственном дефекте этого фермента гомогентизиновая кислота в большом количестве обнаруживается в крови и моче. Моча при стоянии на воздухе, а также при добавлении к ней щелочи становится черной. Это объясняется окислением гомогентизиновой кислоты кислородом воздуха и образованием в ней алкаптона ("захватывающий щелочь"). Гомогентизиновая кислота из крови проникает в ткани — хрящевую, сухожилия, связки, внутренний слой стенки аорты, вследствие чего появляются темные пятна в области ушей, носа, щек, на склерах. Иногда развиваются тяжелые изменения в суставах. Нарушения обмена триптофана. Основной путь метаболизма триптофана приводит к синтезу амида никотиновой кислоты, который играет очень важную роль в жизнедеятельности организма, являясь простетической группой ряда окислительных ферментов — никотинамидадениндинук-леотида (НАД) и его восстановленной формы никотинамидаденин-динуклеотидфосфата (НАДФ). Поэтому при недостаточности никотиновой кислоты и ее амида нарушаются многие обменные реакции, а при значительном дефиците этих веществ развивается пеллагра. Нарушение обмена триптофана может проявиться также в изменении количества образующегося из него серотонина. 87. Мочевина как конечный продукт азотистого обмена. Биосинтез мочевины, его этапы. Связь орнитинового цикла с метаболизмом фумаровой и аспарагиновой кислот. Нарушения синтеза и выведения мочевины. Гипераммониемия. Мочевина — химическое соединение, диамид угольной кислоты. Белые кристаллы, растворимые в полярных растворителях (воде, этаноле, жидком аммиаке). Мочевина является конечным продуктом метаболизма белка у млекопитающих. Производные нитрозомочевин находят применение в фармакологии в качестве противоопухолевых препаратов. Анализ на мочевину входит в биохимический анализ крови. Нормы: 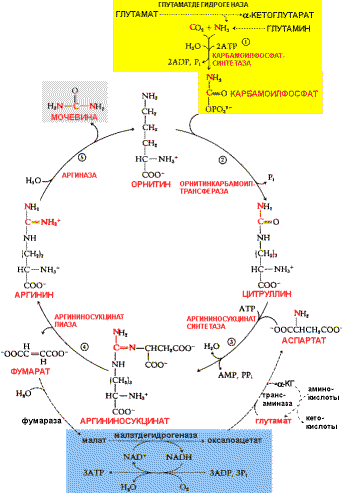
Синтез мочевины — циклический процесс состоит из пяти реакций, катализируемый пятью отдельными ферментами. Суммарное уравнение: СO2+NH3+2H2O+Аспарат → H2N—CO—NH2+Фумарат. При недостаточной активности ферментов орнитинового цикла возникают гипераммониемии— патологические состояния сопровождающиеся повышением концентрации аммиака в крови. 88. Пути обезвреживания аммиака в организме ребенка. Преобладание у детей раннего возраста урикотелического пути обмена аммиака. Причины нарастания аммиака крови. В тканях аммиак находится преимущественно в виде иона аммония NН 89. Биосинтез пиримидиновых нуклеотидов, этапы этого процесса. Оротацидурия. Распад пиримидиновых нуклеотидов. Биосинтез дезоксирибонуклеотидов. Участие УТФ и ЦТФ в обменных процессах. Синтез пуринов и пиримидинов приводит к монофосфатам, соответственно ИМФ (IMP] и УМФ (UMP). Из этих двух предшественников синтезируются все другие нуклеотиды. Синтез пуриновых нуклеотидовосуществляется из инозинмонофосфата[ИМФ (IMP)]. Его основание гипоксантинпревращается в две стадии соответственно в аденин или гуанин. Образующиеся нуклеозидмонофосфаты АМФ(AMP) и ГМФ (GMP) переходят в дифосфаты АДФ(ADP) и ГДФ (GDP) под действием нуклеозидфосфаткинази, наконец, фосфорилируютсянуклеозиддифосфаткиназамидо трифосфатов АТФ(АТР) и ГТФ (GTP). Нуклеозидтрифосфаты служат строительными блоками для РНК (RNA) или функционируют в качестве коферментов. Преобразование рибонуклеотидов в дезоксирибонуклеотиды происходит на стации дифосфатов и катализируется нуклеозиддифосфат-редуктазой. Пути биосинтеза пиримидиновых нуклеотидов: прежде всего исходный УМФфосфорилируется до ди-, а затем трифосфата УТФ. УТФ превращается цитидинтрифосфат-синтазой (CTP-синтаза) в ЦТФ. 2'-Дезоксирибоза, структурный элемент ДНК, не синтезируется в виде свободного сахара, а образуется на стадии дифосфата при восстановлении ρибонуклеозиддифосфатов. Такое восстановление — сложный процесс, в котором участвует несколько белков. Необходимые восстановительные эквиваленты поставляются НАДФН(NADPH). Тем не менее, они не переносятся непосредственно от кофермента к субстрату, а проходит прежде всего через ряд окислительно-восстановительных реакций. На первой стадии тиоредоксинредуктазавосстанавливает с помощью связанного с ферментом флавинадениндинуклеотида небольшой белок, тиоредоксин. При этом дисульфидный мостик в тиоредоксине расщепляется. Образующиеся SH-группы снова восстанавливают каталитически активный дисульфидный мостик в нуклеозиддифосфат-редуктазе(«рибонуклеотид-редуктаза»). Свободные SH-группы являются действенными донорами электронов для восстановления рибонуклеотиддифосфатов. Рибонуклеотид-редуктаза эукариот представляет собой тетрамер, состоящий из двух R1- и R2 -субъединиц. Кроме упомянутого дисульфидного мостика,в ферменте во время реакции образуется тирозин-радикал, генерирующий радикал в субстрате. Последний отщепляет молекулу воды и вследствие этого переходит в радикал-катион. При последующем восстановлении образуется остаток дезоксирибозы и регенерируется тирозиновый радикал. Процесс регуляции рибонуклеотид-редуктазы имеет довольно сложный механизм. Субстратная специфичность и активность фермента контролируются двумя аллостерическими центрами связывания R1-субъединицы. АТФ и дАТФ соответственно повышают и уменьшают активность редуктазы, связываясь с центром а. С центром б взаимодействует другой нуклеотид, изменяющий в результате связывания субстратную специфичность фермента. 90. Распад нуклеиновых кислот. Нуклеазы пищеварительного тракта и тканей. Биосинтез пуриновых нуклеотидов. Источники атомов пуринового кольца. Синтез пуриновых нуклеотидов: аденина и гуанина. Распад пуриновых нуклеотидов, образование мочевой кислоты. Нарушения обмена пуриновых нуклеотидов, подагра, синдром Леша-Нихана. Нуклеазы — большая группа ферментов, гидролизующих фосфодиэфирную связь между субъединицами нуклеиновых кислот. Различают несколько типов нуклеаз в зависимости от их специфичности: экзонуклеазы и эндонуклеазы, рибонуклеазы и дезоксирибонуклеазы, рестриктазы и некоторые другие. Рестриктазы занимают важное положение в прикладной молекулярной биологии. Подагра — гетерогенное по происхождению заболевание, которое характеризуется отложением в разли чных тканях организма кристаллов уратов в форме моноурата натрия или мочевой кислоты. В основе возникновения лежит накопление мочевой кислоты и уменьшение её выделения почками, что приводит к повышению концентрации последней в крови (гиперурикемия). Клинически подагра проявляется рецидивирующим острым артритом и образованием подагрических узлов — тофусов. Чаще заболевание встречается у мужчин, однако в последнее время возрастает распространённость заболевания среди женщин, с возрастом распространённость подагры увеличивается. Синдром Лёша — Нихена — наследственное заболевание, характеризующееся увеличением синтеза мочевой кислоты (у детей) вызванное дефектом фермента гипоксантин-гуанинфосфорибозилтрансферазы, который катализирует реутилизацию гуанина и гипоксантина — в результате образуется большее количество ксантина и, следовательно, мочевой кислоты. 91. Обмен нуклеиновых кислот и нуклеотидов у ребенка. Генетические нарушения, связанные с наследственными заболеваниями. |