Физическая химия конспект лекций. Конспект лекций Введение
 Скачать 1.05 Mb. Скачать 1.05 Mb.
|

|
Ртутно-сульфатные электроды SO42 –/Hg2SO4, Hg аналогичны каломельным с той лишь разницей, что ртуть здесь покрыта слоем пасты из Hg и закисного сульфата ртути, а в качестве раствора используется H2SO4. Потенциал ртутно-сульфатного электрода при 25 oС выражается уравнением: Хлорсеребряный электрод представляет собой систему Cl–/AgCl, Ag, а его потенциалу отвечает уравнение: ECl– /AgCl, Ag = E0Cl–/AgCl, Ag –b lg aCl– или при 25 оС: ECl–/AgCl, Ag = 0,2224 – 0,0592 lg a Cl–. 9. Электроды сравнения Электроды сравнения – электроды, используемые при измерении электродных потенциалов в паре с используемым электродом. Электродный потенциал – скачок потенциала на границе металл-раствор. Он определяется: природой металла, раствора, концентрацией, температурой. Для сравнения электродных потенциалов нужны стандартные условия: t = 25 °С = 298 К; Р – 1 атм, одномолярный раствор. Абсолютное значение электродного потенциала измерить нельзя. Поэтому измеряют разность потенциалов между данным электродом и электродом сравнения, потенциал которого принимают равным нулю. Часто используют водородный электрод, изготовленный из губчатой платины с сильно развитой поверхностью (платиновая чернь), опускают в раствор H2SO4 – серной кислоты с активностью ионов водорода, равной единице. При этом через раствор пропускается газообразный водород под давлением, который затем адсорбируется платиной. Относительно потенциала водородного электрода все металлы располагают в ряд напряжений, установленный электрически Н. Н. Бекетовым, взаимному вытеснению металлов в зависимости от величины и знака стандартного электродного потенциала. Существуют и другие электроды сравнения: каломельный, хлорсеребряный и другие, в зависимости от различных методов. Конструктивное оформление электрода сравнения разнообразно. Например, для полярографического метода электроды сравнения должны иметь большую поверхность во избежание поляризации их при работе под током. ЛЕКЦИЯ № 13. Электрохимическая кинетика 1. Основные кинетические характеристики и методы их расчетов i0 – ток обмена – кинетическая характеристика равновесия между электродом и раствором при равновесном значении электродного потенциала. Токи обмена относят к 1 см2 поверхности раздела электрод-раствор. α– коэффициент переноса заряда – характеризует степень влияния электрического поля электрода на энергию активации электрохимической стадии и определяет симметрию катодного и анодного процессов, зависит от формы потенциальных кривых. α≈ 0,5. При одном и том же отклонении потенциала электрода от равновесного значения скорости реакции результативная плотность тока будет тем больше, чем выше i0. Ток обмена i0 зависит от природы электрохимической реакции, материала электрода и состава раствора. Константа скорости – скорость реакции при единичных концентрациях. Скорость прямой реакции: где k – константа, зависящая от свойств системы и способа выражения скорости процесса; Cox– концентрация реагирующих частиц; Ea– энергия активации разряда в отсутствии скачка потенциала между металлом и раствором. Скорость обратной реакции где CRed – концентрация частиц Red (восстановление продуктов); Еa– энергия активации реакции ионизации при скачке потенциала между металлом и раствором, равным нулю. Энергия активации электрохимического процесса зависит от величины электродного потенциала, природы ее непосредственных участников и электрода. Энергия активации при постоянном перенапряжении η – эффективная энергия активации. Если энергия активации не зависит от перенапряжения, то ее появление замедляет диффузию. Метод расчета величин αи i0 основан на явлении редоксикинетического эффекта заключается в том, что при наложении переменного тока на электрод его потенциал смещается в ту или иную сторону на некоторую величину от первоначального значения. Это смещение – редоксикинетический потенциал ψ. Связь редоксикинетического потенциала ψ с кинетическими параметрами а и i0 такая: если наложить переменный ток на электрод, находящийся в равновесии с соответствующими ионами в растворе, то за время катодного полупериода он окажется заполяризованным катодно, причем зависимость между ηи iпри условии замедленности стадии разряда будет передаваться уравнением 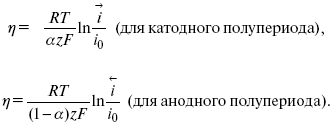 При достаточном удалении от состояния равновесия 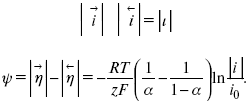 Из последнего выражения следует, если α= 0,5, то ψ = 0, чем сильнее αотклонена от 0,5, тем больше ψ. Энергия активизации – энергия, представляющая собой минимальную энергию, достаточную для осуществления акта химической реакции. 2. Уравнения электрохимической кинетики, пределы их применимости 1-й закон Фарадея устанавливает прямую пропорциональность между количеством прошедшего через систему электричества и количеством прореагировавшего вещества. Δm = kэJt = kэq, (1) где Δm – количество прореагировавшего вещества; k – коэффициент пропорциональности; q – количество электричества, равное произведению силы тока I на время t. Если q = Jt = 1, то Δm = kэ– количество вещества, прореагировавшего в результате протекания единицы количества электричества. kэ– электрохимический эквивалент. 2-й закон Фарадея устанавливает связь между количеством прореагировавшего вещества при пропускании данного количества электричества и его природой. По этому закону, при постоянном количестве прошедшего электричества массы прореагировавших веществ относятся между собой, как их химические эквиваленты А: Если количество электричества равно F, числу Фарадея, то Δm1 = Fkэ1 = A1, Fkэпри q = 1F, то Уравнение (3) позволяет объединить оба закона Фарадея в виде одного общего закона, по которому количество электричества (1F = 96500k) всегда изменяет электрохимически массу любого вещества, независимо от его природы. Законы Фарадея – основные законы электролиза, согласно которых, количество вещества, выделившегося при электролизе, прямо пропорционально его химическому эквиваленту и количеству прошедшего электричества. Уравнение Нернста Е0 – равновесный стандартный потенциал. где С0 – стандартная концентрация раствора; С – любая концентрация в нестандартных условиях, С = С0 x Е = Е0 , т. е. в стандартных условиях С = C0 = 1 моль. Для окислительных веществ 1-й закон Фика: где dc/dx – градиент концентрации; s – площадь, через которую происходит диффузия. Δ – коэффициент диффузии cм2 x c-1, показывает число частиц, продиффундировавших за 1 с через поперечное сечение раствора площадью 1 см2, dt – время диффузии, dm – число продиффундировавших частиц. где Тк – коэффициент внутреннего трения; D – коэффициент диффузии. Первый закон Фика относится к процессу стационарной диффузии, сходен с закономерностями переноса тепла из электричества. Если диффузионный поток не изменяется с течением времени, это называется стационарной диффузией. Диффузия – самопроизвольно протекающий в системе процесс выравнивания концентрации молекул, ионов, частиц под влиянием теплового хаотического движения. Основное уравнение электрохимической кинетики ik = ia = i0, где i0 – ток обмена, (окислительно-восстановительные реакции). При катодной поляризации на электроде через систему протекает ikпреимущественно, если поляризация не слишком велика, то суммарная скорость процесса равна: i = ik – ia, для реакции (1) катодные и анодные токи будут равны: 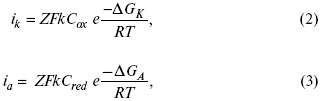 где Z – количество электронов, участвующих в реакции; F – число Фарадея; к – const скорости; Сox, Cred – концентрация окислительной и восстановленной форм реагентов; ΔGK – энергия активации катодного процесса; ΔGA – энергия активации анодного процесса. Энергия активации зависит от величины накладываемого потенциала, в то же самое время эта энергия распределяется между прямой и обратной реакцией в соответствии с коэффициентом переноса – а, т. е. υ = υпр – υоб. Коэффициент переноса α– доля энергии электрического поля в ДЭС, которая приходится на прямую и обратную реакции. α– коэффициент переноса для катодной реакции; (1 – α) – для анодного процесса (коэффициент переноса). ΔGk = ZFE α, (4) ΔGA = ZFE(1 – α) (5) С учетом уравнений (4), (5) уравнения (2), (3) примут следующий вид: 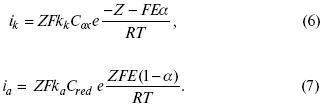 Различие знаков у электрона объясняется тем, что катодная поляризация («–») ускоряет прямую реакцию и замедляет обратную реакцию. 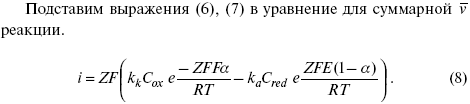 Введем в уравнение (8) плотность тока обмена – i0. Вместо потенциала введем перенапряжение: полное уравнение поляризационной кривой. Вывод из уравнения (10): 1) при равновесном потенциале, когда ток равен нулю, уравнение (10) преобразуется в уравнение Нернста: 2) при малых величинах η: 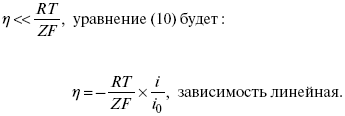 При сдвижении потенциала от равновесного (59 mВ); 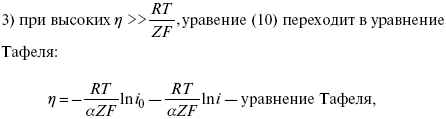 η = a + b ln i– уравнение Тафеля в простом виде при замедлении стадии переноса заряда. Величина i0 (тока обмена) и α(коэффициента переноса) – основные кинетические параметры стадии переноса заряда (q). Они могут быть определены из экспериментальных измерений, для этого на исследуемом электроде снимают зависимость η– i или Ei – i. Поляризационная кривая судит о коррозионной стойкости металлов. Перестраиваем поляризационную кривую в координаты: 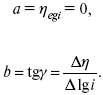 Определяем const а и bв уравнении Тафеля, определяем b: Из коэффициента bнайдем а, после подставим в а и найдем i0. Перенапряжение Н2 (водорода). Источник выделения Н2 – Н2SO4 →Н++ НSO4– Источник выделения Н2 – Н2О → Н++ ОН-. В рН < 7 Н2 выделяется по реакции. 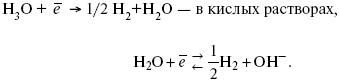 Н2 всегда выделяется в потенциалах более отрицательных, чем равновесный потенциал, то есть с перенапряжением. Суммарный процесс выделения водорода состоит из следующих стадий: 1) доставка к поверхности катода реагирующих частиц Н3О+; 2) разряд Н3О+ с образованием Надс 3) удаление выделяющегося Надс с поверхности электрода может происходить тремя путями: а) каталитическая рекомбинация где Кat – материал катода; б) электрохимическая десорбция – удаление Н2 происходит на уже адсорбированных атомах в) эмиссия включает две стадии: 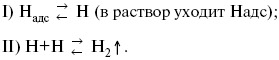 Для Pt замедлена стадия а), для других металлов (Hg, Pb) – стадия разряда, Н+ – самый подвижный. 3. Кинетические особенности электроосаждения металлов и сплавов Процесс электроосаждения металлов, сплавов протекает через последовательность стадий: диффузия катионов металлов к поверхности электрода из объема раствора, вхождение катионов в ДЭС (двойной электрический слой), потери сольватной оболочки, переход катионов в состояние адсорбции атома, полный перенос заряда с поверхности электрода на разряжающийся ион или адсорбированного атома (ад. атома) и образование зародышей металлов, рост зародышей и заполнение поверхности новой фазы в виде сплошного слоя, рост слоя осадка в толщину. Процесс электровыделения не зависит от состояния поверхности электрода, в частности, большое влияние на η(перенапряжение) процесса оказывает концентрация вакансий на поверхности электрода. Кристаллическая решетка каждого металла содержит определенное количество равновесных вакансий (свободных незанятых узлов в кристаллической решетке). Наличие таких пустот в структуре поверхностного слоя облегчает образование ад.атомов, так как в местах вакансий имеет место более сильное энергетическое воздействие кристаллической решетки на образующиеся атомы новой фазы. После заполнения этих активных мест начинается рост зародышей, т. е. образование скоплений атомов, которые постепенно заполняют всю поверхность. С другой стороны, скорость реакции электровыделения металлов зависит от состояния катионов этого металла в растворе. В растворе катионы находятся в сольватированном виде или в виде комплексов. Разрушение сольватной оболочки происходит на границе плотного слоя Гельмгольца с диффузной частью ДЭС. Таким образом, реакции разряда, протекающие в плотном слое Гельмгольца, энергетически возможны только в том случае, если ионы металла преодолевают потенциальный барьер. Высота потенциального барьера, т. е. величина энергии, которую ионам в растворе нужно преодолеть, чтобы попасть из раствора в плотный слой Гельмгольца, может быть различной, и определяется она природой растворителя, лигандов, прочностью связей в комплексах. Пример: 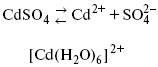 (заряд комплекса не меняется, так как молекула нейтральна). Сама стадия переноса зарядов также протекает стадийно 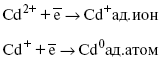 Анионные комплексы наиболее прочные, и последняя стадия состоит из процесса распада до свободного иона на поверхности электрода в слое Гельмгольца. Это обусловлено тем, что анионы, обладающие высокой поверхностной активностью, связываются с поверхностью электрода и оказывают влияние на распределение заряда в ДЭС. Итак, помимо диффузии в объеме раствора, диффузии ад. ионов, ад. атомов по поверхности, стадий переноса заряда, образования зародышей и роста зародышей в сплошной слой (стадия кристаллизации), на скорость реакции могут оказывать влияние также реакции разложения комплексов в растворе, гомогенная химическая стадия, предшествующая стадии разрядов, и гетерогенная химическая стадия на поверхности электродов. Скорость реакции определяется концентрацией потенциал-определяющих частиц в растворе; концентрация потенциал-определяющих частиц зависит от состояния ионов. Состояние ионов в растворе определяется энергией взаимодействия с молекулами растворителя и лигандами. Потенциал электрода определяется активностью ионов раствора. В случае твердых металлических электродов активность самого металлического электрода не сказывается на длительности процесса и на величине скорости потенциала электродов (принято считать α твердой фазы = 1). Если металл растворен в ртути (Hg), то в этом случае i зависит от α металла фазы в матрице электрода 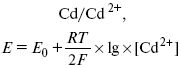 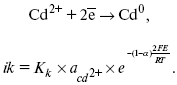 |