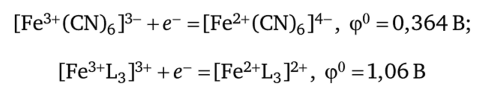заттар құрылысы мен құрамы. лекция посл. Лекция 1 Введение. Предмет аналитической химии
 Скачать 0.52 Mb. Скачать 0.52 Mb.
|

=Fe2+, ф°=0,771В. Той же редокс-паре в комплексах соответствуют уже другие величины стандартного ОВП:
. Ионы Со2+ образуют с тиоцианат-ионами комплексы [Co(SCN)4]2_ синего цвета:
, SO3-, 520з“, AsO§-. Раствором перманганата калия КМп04 в сернокислой среде окисляются перечисленные выше анионы, а также С20^_ (при нагревании раствора), В r_, I-, CN-, SCN- и СЬ, который реагирует медленно. Эти реакции используют для открытия соответствующих анионов.Свойства и характеристика растворителей. Водные и неводные растворы сильных и слабых кислот и оснований. Вычисления рН растворов кислот, оснований, смеси кислот и оснований. Состояние ионов элементов в растворах. Буферные растворы, используемые в химическом анализе, их состав, свойства, расчёт рН, буферная ёмкость, область буферирования, применение в аналитической химии. Использование реакций гидролиза в аналитической химии. Вода представляет собой слабый электролит, ионизирующий по уравнению: H2O = H+ + OH- Реакции ионизации воды сопровождаются переносом протона от одной молекулы воды к другой. Такие процессы называют процессами автопротолиза. Наряду с ионами анализируемого вещества всегда находятся ионы Н+ и ОН-. Константа ионизации воды будет: Кравн.= Так как вода находится в виде неионизированных молекул, величину [H2O] можно считать практически постоянной. Приняв количество молей в одном литре равной: [H2O] = получим константу автопротолиза КW, или ионное произведение воды: Кравн.= Степень ионизации воды мала, так при 250 С в 1 л воды распадается на ионы лишь 10-7 моль воды. Так как из каждой молекулы воды получается по одному ионы Н+ и ОН-, концентрации их в чистой воде равны: [H+]=[OH-]=10-7моль/л (при 250С). Константу автопротолиза для удобства представляют в виде показателя константы автопротолиза воды, представляющего обратный логарифм константы автопротолиза: рКW = -lgKW = -lg10-14 = 14 Концентрацию водородных и гидроксильных ионов обозначают в виде рН и рОН: рН = -lg[H+]; pOH = - lg[OH-]; pH + pOH = 14 = pKW. В нейтральном водном растворе, т.е. без присутствия посторонних соединений: [H+] = [OH-] = Для кислых растворов: [H+] > [OH-]; т.е. [OH-] > 10-7 моль/л Для щелочных (основных) растворов: [H+] < [OH-]; тогда [H+] < 10-7. Расчет рН В аналитической химии важно знать концентрации всех частиц в растворе кислоты или основания после установления равновесия, в частности концентрацию ионов водорода (рН раствора). 1. Раствор сильной кислоты или сильного основания. Протолитические равновесия: НАn + Н2О → Н3О+ + Аn-; В + Н2О → ВН+ + ОН- Полностью смещены вправо, поэтому: [H+] = CHAn, и [OH-] = CB, откуда: рН = -lg [H+] = -lg CHAn и рОН = -lg[OH-] = -lgCB 2.Раствор слабой кислоты и слабого основания. В растворе слабой кислоты устанавливается равновесие: HA = H+ + An- K = Тогда [H+]2 = K ·CHan; [H+] = Вывод уравнения для вычисления рН слабых оснований основан на рассуждениях подобных для слабых кислот, т.к. слабые основания ионизируют не полностью: BOH = B+ + OH- То K = При этом [OH-]=[B+]<<1, а [BOH]=CBOH Тогда [OH-]2=K· CBOH, [OH-]= pH=14-1/2lgK+lg CBOH Буферными растворами называют растворы веществ, способные сохранять постоянное значение рН при добавлении к ним небольших количеств сильных кислот или оснований, а также при разбавлении. Буферный раствор содержит либо слабую кислоту и ее соль (кислотные буферные системы), либо слабое основание и ее соль(основные буферные системы). Например, ацетатный буфер – это раствор уксусной кислоты и ацетата натрия, аммиачный буфер – раствор аммиака и хлорида аммония. Иногда в качестве буферного раствора применяют смесь кислой и средней солей, например карбонатный буфер – NaHCO3 + Na2CO3. Буферные системы характеризуются двумя параметрами: значениями создаваемого рН и буферной емкостью. Значение рН в буферных системах определяется величиной констант ионизации слабой кислоты или слабого основания соответственно, а также в кислотных системах соотношением концентрации слабой кислоты CA и ее соли CC; в основных системах – соотношением концентрации слабого основания CB и его соли CC. Равновесные состояния в буферных системах можно рассматривать как равновесие процесса ионизации соответствующих слабых кислот или оснований. Если обозначить концентрацию кислоты [HA] = CHA, а аниона [A-], как концентрацию соли СС (аналогично [B] = CB и [BH+] = CC), то из констант равновесия можно получить формулы расчета рН в буферных растворах: Кислотные буферные системы: Основные буферные системы: HA → H+ + A- B + H+ → BH+ KA = При [HA] = CHA и [A-] = Cc при [B] = CB [BH+] = Cc и KBH+ = Kw/KB [H+] = рН = рКА + lg При равенстве концентраций кислоты и соли СС = СНА (и основания и соли СС= СВ) формулы упрощаются: рН = рКА и рН = рКW – рКВ. Буферная емкость системы описывается количеством молей кислоты или основания, которые при добавлении изменяют на единицу рН 1 дм3 буферного раствора. Буферные растворы широко используются в аналитической химии. В качественном анализе многие процессы осаждения труднорастворимых электролитов проходят при определенном значении рН раствора. Например, при открытии ионов магния с помощью моногидрофосфата натрия применяется аммонийная буферная смесь, создающая рН ≈ 8,5. Буферные растворы широко применяются и в количественном анализе. Титриметрическое определение многих металлов с помощью трилона Б проходит при определенном рН (8-10). Необходимая среда создается также с помощью аммонийной буферной смеси (NH4OH + NH4CI). Водные растворы многих солей имеют кислую или щелочную реакцию. Как, известно, кислотность или щелочность раствора в водной среде зависит от избытка в растворе ионов водорода или гидроксила. Наличие в растворе избытка Н+ или ОН- - ионов объясняется взаимодействием ионов соли с водой. Например: NH4+ + HOH = NH4OH + H+ или CH3COO- + HOH = CH3COOH + OH- Реакция взаимодействия ионов соли с водой, приводящая к образованию слабого электролита, называется гидролизом соли. Каждую соль можно представить себе образованной основанием и кислотой. Кислоты и основания могут быть сильными и слабыми электролитами. Следовательно, по этому признаку соли можно разделить на четыре типа: соли, образованные катионом сильного основания и анионом слабой кислоты (CH3COONa, NaCN). Такие соли подвергаются обратимому (неполному) гидролизу, в растворе постепенно будут накапливаться ионы OH- и поэтому раствор соли будет иметь щелочную среду. соли, образованные катионом слабого основания и анионом сильной кислоты (NH4CI, ZnCI2). Такие соли подвергаются обратимому гидролизу, в растворе накапливаются ионы Н+ и раствор соли будет иметь кислую среду. соли, образованные катионом слабого основания и анионом слабой кислоты (CH3COONH4, (NH4)2CO3). В первом примере процесс гидролиза будет обратимым, и раствор соли будет иметь нейтральную среду. Во втором случае, результате такого гидролиза получаются вещества, одно из которых малорастворимое и выпадает в осадок, а другое является нестойким или летучим и гидролиз протекает полностью (необратимый гидролиз) и реакция будет слабощелочной. соли, образованные катионом сильного основания и анионом сильной кислоты (KCI, Na2SO4). При растворении в воде таких солей не происходит связывания ни ионов Н+, ни ионов ОН- воды, следовательно они гидролизу не подвергаются. Для большинства солей процесс гидролиза обратим. Состояние гидролитического равновесия характеризуется степенью гидролиза. Степень гидролиза соли (h) – отношение концентрации гидролизованной соли (СS) к ее общей концентрации (CO): h = Степень гидролиза связана с константой ионизации образующегося при гидролизе слабого электролита. Чем меньше константа ионизации слабого электролита, тем выше степень гидролиза. Константа и степень гидролиза связаны между собой. Учитывая, что гидролиз является разновидностью реакций протолиза, или ионизации, можно применить закон разведения Оствальда: КS = Откуда hS = При проведении анализа необходимо знать значение рН в растворах гидролизующихся солей. Формулы расчета рН можно вывести из формул констант равновесия. Для соли слабой кислоты и сильного основания в воде находим: [H+] = Для соли сильной кислоты и слабого основания: [H+] = Для соли слабой кислоты и слабого основания: [H+] = Реакция гидролиза применяется в аналитической химии для обнаружения отдельных ионов, регулирования кислотности и щелочности анализируемых растворов, разделения ионов при качественном систематическом анализе. Процесс гидролиза солей оказывает влияние на проведение количественного анализа. Например, при определении слабых кислот с помощью растворов оснований, рН точки эквивалентности вследствие гидролиза образующейся соли становится больше 7, что учитывают в процессе анализа. Подавление гидролиза осуществляют, добавляя к растворам солей слабых кислот и сильных оснований избыток щелочи, к растворам солей слабых оснований и сильных кислот – избыток сильной кислоты. Равновесие реакции гидролиза при этом сдвигается в сторону образования соли, и гидролиз подавляется. Контрольне вопросы Напишите уравнение диссоциации воды. Что называется ионным произведением воды? Какие растворы называются кислыми, нейтральными, щелочными? Как рассчитывают рН сильных и слабых кислот и оснований? Какая реакция называется реакцией гидролиза? Какие соли могут подвергаться гидролизу? Что показывает степень гидролиза, и от каких факторов она зависит? Лекция№9. Гетерогенные системы в аналитической химии. Основные типы реакций, применяемые в аналитической химии (кислотно-основные, окисления-восстановления, комплексообразования, осаждения). Состояние ионов в растворах. Одним из основних законов химии является закон действующих масс. Он описывает взаимосвязь между концентрацией реагирующих веществ и скоростью химической реакции. Если взаимодействующие вещества означить как А и В, а продукты реакции – как С и Д, то обратимая реакция между А и В выражается в простейшем виде химическим уравнением: nА + m В = cС +d Д Скорость V1 прямой химической реакции пропорциональна произведению концентраций реагирующих веществ в степенях, равных коэффициентам реакции: V1 = K1 [A]n·[B]m Скорость V2 обратной реакции равна: V2 =K2 [C]c · [D]d К – коэффициент пропорциональности; квадратными скобками обозначена концентрация веществ, в моль/л. Коэффициент пропорциональности равен скорости реакции при концентрациях, равных 1 моль/л. Опыт показывает, что скорость химической реакции зависит от температуры, при повышении температуры скорость большинства реакций увеличивается, т.к. увеличивается скорость движения молекул, а следовательно число столкновений между ними. Также скорость реакции зависит и от природы реагирующих веществ. В первый момент протекания реакции концентрации исходных веществ имеют наибольшее значение и скорость прямой реакции велика. При взаимодействии их концентрация падает и скорость уменьшается. При появлении в сисеме продуктов реакции и возрастании их концентрации увеличивается скорость обратной реакции. В момент равенства скоростей прямой и обратной реакций наступает химическое равновесие: V1 = V2 K1[A]n · [B]m = K2 [C]c · [D]d ; При химическом равновесии отношение произведения концентрации продуктов реакции к произведению концентрации исходных веществ есть величина постоянная, называемая константой химического равновесия. Она не зависит от концентрации реагирующих веществ и остается постоянной величиной при постоянных температуре и давлении. Уравнение (1) справедливо только для идеальных растворов, к которым можно отнести разбавленные растворы слабых электролитов. В реальных растворах концентрация ионов не соответствует их действительной химической активности, которая значительно ниже. Константа равновесия химической реакции, учитывающая эффективные концентрации или активности ионов в растворе имеют вид: КТ = и называется термодинамической константой равновесия. При неизменных условиях химическое равновесие может сохраняться как угодно долго. Изменяя условия, можно нарушить это равновесие. Сдвиг равновесия может быть вызван изменением концентрации одного из веществ, изменением температуры и давления. Общую формулировку влияния изменений условий на равновесие системы дает принцип смещения равновесия, сформулированный Ле-Шателье в 1884 г.: Если изменить одно из условий, при которых система находится в равновесии, и, таким образом, нарушить равновесие, то в системе возникают процессы, которые ведут к восстановлению равновесия. Константы равновесия легко позволяют определить направление химической реакции. Исходя из закона действующих масс, прямая реакция преобладает при К> 1, обратная – при К < 1. На основании константы равновесия можно определить применимость реакции для анализа. Чем больше константа равновесия, чем больше образуется продуктов реакции и меньше остается исходных веществ. Реакции в водных растворах представляют собой реакции ионов. Поэтому важно знать, в какой мере различные электролиты распадаются в растворе на ионы. Число, показывающее, какая часть от общего количества растворенного электролита распадается на ионы, называется степенью электролитической диссоциации или ионизации (α): α = Степень диссоциации выражается или в десятичных дробях или, чаще, в процентах. В зависимости от степени электролитической диссоциации кислот, оснований и солей различают сильные, средние и слабые электролиты. Степень диссоциации электролитов зависит от природы растворителя, концентрации раствора, природы растворителя, температуры. Рассмотрим ионизацию слабых электролитов. Причиной ионизации является притяжение, испытываемое молекулами уксусной кислоты со стороны молекул воды. Молекулы воды построены несимметрично, вследствие этого «электрические центры тяжести» отрицательных и положительных зарядов в молекуле воды не совпадают, а находятся в двух различных точках пространства. Такие молекулы называются электрическими диполями, они создают силу электростатического притяжения, стремящиеся как бы разорвать молекулу электролита на ионы и тем самым ослабить связь между ними. В результате ионизации образуются не собственно ионы, а соединения их с молекулами растворителя и если растворителем является вода, то такие соединения называются гидратами. CH3COOH + (n+1)H2O = H3O+ + [CH3COO·nH2O]- Следовательно, ионизацию надо рассматривать не как чисто физический процесс, а как химическое взаимодействие растворяемого вещества с растворителем. Это Д.И.Менделеев положил в основу своей гидратной теории растворов. Процесс ионизации обратим, не учитывая гидратную воду, процесс ионизации уксусной кислоты можно представить следующим образом: CH3COOH → H+ + CH3COO- По закону действия масс: Ск и Са – концентрации катиона и аниона; СМ – концентрация неионизированных молекул данного электролита. Величина К называется константой ионизации электролита. Она характеризует склонность электролита к распаду на ионы. Если раствор содержит С моль/л уксусной кислоты, а степень ионизации ее равна α, то число ионизированных молей CH3COOH будет равно Сα, отсюда: СК = СА = Сα Концентрацію неионизированных молекул уксусной кислоты полу чим: СМ = С – Сα = С(1 - α) Подставляя полученные значения СК, СА и СМ в уравнение (), получим: Это уравнение выражает закон разбавления Освальда, который устанавливает зависимость между степеню ионизации слабого электролита и его концентрацией. Если электролит слабый и раствор его не слишком разбавленный , его степені ионизации мала и величина (1- α ) мало отличается от единицы, то Сα2 ≈ К и α = Отсюда видно, что степень ионизации должна возрастать с разбавлением раствора. Состояние сильних электролитов в рас творах. Ионизация сильних электролитов закону действия масс не подчиняется, в отличие от слабих электролитов сильне электролиты констант диссоциации не имеют. Согласно теории сильных электролитов введенная в 1923 г. Дебаем и Гюккелем сильне электролиты в растворах практически полностью ионизированы на ионы. Для оценки способности сильних электролитов к химическим взаимодействиям употребляется термин активность. Под активностью иона (а) понимают ту эффективную, кажущуюся концентрацию его, соответственно которой он действует в химических реакциях. Отношение активности к действительной концентрации иона называется коэффициентом активности: ff = Дальнейшим развитием теории сильных электролитов явился закон ионной силы, который гласит, что коэффициент активности данного электролита один и тот же во всех разбавленных растворов, имеющих одинаковую ионную силу: μ = где С1, С2, …Сn – концентрации отдельных ионов в растворе, моль/л; z1, z2, …..zn – их заряды Математическую связь между ионной силой раствора и коэффициентом активности выражается уравнениями. Для разбавленных растворов (0,01 – 0,05 н): -lg f = 0,5 z2 Для более концентрированных растворов (0,1 – 0,5 н.): -lg f = 0,5 z2 Контрольные вопросы Что следует понимать под скоростью химической реакции? В каких единицах измеряется скорость химической реакции? Назовите факторы влияющие на скорость реакции Каков физический смысл константы скорости реакции? Какие реакции называются необратимыми? Дайте формулировку понятия константы химического равновестия Что называется смещением равновесия? Что такое степень электролитической диссоциации? Какие электролиты называются слабыми, сильными? Что такое активность? Лекция № 10. Применение амфотерности и гидролиза в химическом анализе. Состав любого гидроксида можно выразить формулой R(OH)x, где R — центральный ион, а ОН- — гидроксид-ион. Отнесение гидроксида к классу оснований или кислот зависит от типа диссоциации его молекул в водном растворе. Полярные молекулы при растворении в воде диссоциируют по месту наиболее полярной связи. Например, в молекуле серной кислоты наиболее полярной является связь О—Н и диссоциация происходит с отщеплением катионов Н+. В молекуле гидроксида калия наиболее полярная связь О—К, и соединение диссоциирует с образованием гидроксид-ионов ОН |
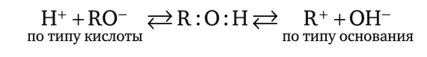
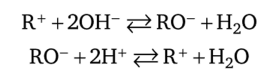
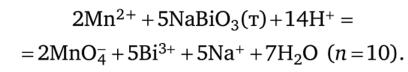
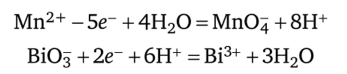
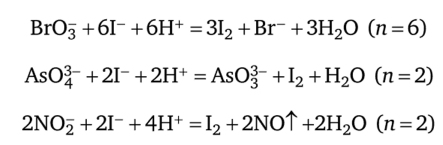
Некоторые из методов количественного анализа основаны на использовании окислительно-восстановительных реакций. Так, при гравиметрическом определении железа (II) его предварительно окисляют до железа(Ш), прибавляя концентрированную азотную кислоту, после чего осаждают раствором аммиака в форме гидроксида железа(Ш), который затем прокаливают до оксида Fe203 и взвешивают. При этом протекают реакции:
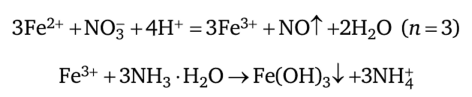
Аналогично гравиметрически определяют иногда никель (II) в его соединениях, окисляя бромной водой никель (II) до никеля(III) с последующим осаждением гидроксида нике- ля(Ш), прокаливанием его до оксида Ni203 и взвешиванием последнего.
Один из разделов количественного титриметрического (объемного) анализа целиком основан на применении окислительно-восстановительных реакций — это окислительно-восстановительное титрование (оксидиметрия, редоксметрия). К наиболее распространенным методам редоксметрии относятся перманганатометрия, иодиметрия и иодоме- трия, броматометрия, нитритометрия, дихроматометрия, цериметрия. Все они используются в анализе различных биологических объектов.
Для определения галогенов, серы, фосфора в органических соединениях используют метод сжигания образца — озоление (почвы, растений) в атмосфере кислорода, при котором протекают глубокие окислительно-восстановительные процессы. При этом продукты сгорания (окисления) растворяют (сорбируют) в поглощающей жидкости, после чего анализируют образовавшийся раствор.
.
Контрольные вопросы
1. .Окислительно-восстановительное равновесие.
2.Окислительно-восстановительный потенциал.
3.Окислительно-восстановительные реакции и составление уравнений реакций.
4.Влияние условии реакции на окислительно-восстановительные процессы.
Лекция №12. Значение коллоидных систем в химическом анализе.
Признаки образования коллоидных растворов. Очень часто при выполнении химического анализа в процессе осаждения, фильтрования и промывания некоторых осадков образуются коллоидные системы (коллоидные растворы,
Явление прохождения коллоидных частиц через фильтр с образованием мутного фильтрата можно заметить при фильтровании и промывании осадков сульфатов катионов второй и осадков сульфидов и гидроокисей катионов третьей, четвертой и пятой аналитических групп, а также галогенидов серебра и др.
Гели. Коллоидные частицы под влиянием различных факторов соединяются в более крупные агрегаты. Процесс укрупнения коллоидных частиц называют коагуляцией. Свертывание коллоидных частиц приводит в конце концов к образованию сплошной и рыхлой массы, называемой гелем.
При действии на осадки некоторых реагентов (пептизаторов) наблюдается явление, обратное коагуляции, называемое пептизацией. В процессе пептизации осадок переходит в золь.
Коллоидным частицам присущи характерные свойства и в том числе адсорбционные свойства (см. книга 2, гл. IV, § 6).
Предупреждение образования золей. Во избежание образования золей при выполнении аналитических работ рекомендуется:
1. В процессе осаждения осадков прибавлять лишь небольшой избыток осаждающего реактива. Это не только способствует уменьшению растворимости осадков, но и предупреждает переход малорастворимого соединения в золь.
2. Вести осаждение из разбавленных растворов при нагревании и перемешивании.
3. При осаждении и промывании осадков добавлять необходимые электролиты.
4. Избегать разбавления водой растворов, находящихся над осадком.
Использование коллоидных систем в анализе. Способность некоторых соединений образовывать золи используют в химическом анализе для повышения чувствительности реакций. Так, некоторые гидроокиси, особенно при малых концентрациях ионов, образуют плохо видимые коллоидные осадки. Видимость этих осадков может быть значительно усилена, если использовать адсорбционные свойства коллоидных части . Для этого к раствору, в котором происходит осаждение гидроокиси, прибавляют какое-либо красящее вещество. Например, при осаждении гидроокиси магния добавляют
Явление -адсорбции коллоидными частицами широко используется в анализе, например для соосаждения
Другим примером использования свойств коллоидных систем может служить адсорбция суспензией метаоловянной кислоты значительных количеств фосфорной кислоты, чем пользуются для количественного отделения фосфат-ионов, осложняющих ход анализа смеси катионов (см. гл. VI, § 22).
Коллоидные системы широко применяются в количественном анализе (см. книга 2).
Контрольные вопросы
1. .Коллоидные системы.
2.Явление коагуляции и пептизации.
3.Соосаждение
Лекция № 13. Применение реакции образование комплексных соединений в аналтической химии.
Осаждение катионов и анионов из растворов. Катионы калия К+ осаждают с помощью гексахлороплатината натрия Na2[PtCl6] или платинахлористоводородной кислоты:
Осаждение катионов калия можно также проводить с помощью гексанитрокобальтата(Ш) натрия Na31 Co(N02)6 |:
Еще менее растворим в воде гексанитрокобальтат(Ш) калия и серебра(1) K2Ag[Co(N02)6], осаждающийся из водных растворов в виде оранжево-желтого осадка.
Осаждение катионов аммония NH^ можно осуществить растворами гексахлороплатината натрия Na2[PtCl6] или платинахлористоводородной кислоты H2[PtCl6] (как и катионов калия):
Висмут(Ш) осаждают из водных растворов в форме комплекса [Cr(NH3)6] [BiCl6].
Аналогично можно осадить из водных растворов сурьму(У) в виде устойчивого и весьма малорастворимого комплекса [Сг(еп)3] |SbS4], где еп — молекула этилендиамина. Это очень удобный и быстрый метод определения полумикроколичеств сурьмы.
Растворение осадков. Для растворения осадка хлорида серебра AgCl используют реакцию образования растворимого комплекса [Ag(NH3)2]Cl
Обработка щелочами при нагревании осадка сульфата свинца PbS04 приводит к его растворению вследствие образования растворимого комплекса [РЬ(ОН)4]2-:
в ионной форме
Разделение ионов путем дробного осаждения или дробного растворения. Ионы кадмия Cd2+ и цинка Zn2+, обладающие сходными свойствами, можно разделить дробным осаждением с последующим их определением. Для этого в раствор, содержащий смесь катионов кадмия и цинка, вводят тиокарбамид SC(NH2)2 и затем комплекс хрома(Ш) [Cr(SCN)4]. Образующийся растворимый тиокарбамидный комплекс кадмия
где L — молекула тиокарбамида, осаждается в форме малорастворимого осадка красного цвета с комплексом хрома
Осадок кадмиевого комплекса можно отделить от маточника, в котором остаются катионы цинка.
Другой пример — отделение серебра из смеси хлоридов серебра и ртути: AgCl + Hg2Cl2. Осадок, состоящий из этой смеси, обрабатывают раствором аммиака. Серебро переходит в раствор вследствие образования растворимого комплекса [Ag(NH3)2]Cl
Хлорид ртути (I) Hg2Cl2 остается в осадке, который постепенно темнеет вследствие выделения металлической ртути при реакции хлорида ртути(1) с аммиаком.
Аналогично можно перевести в раствор из осадков гидроксидов катионы Со2+, Ni2+, Cu2+, Cd2+, Hg2+ в форме растворимых аммиачных комплексов, тогда как гидроксиды магния, марганца, железа, сурьмы, висмута остаются в осадке, поскольку не образуют растворимых аммиакатов.
Определение ионов по изменению окраски раствора вследствие образования окрашенных комплексов. Многие комплексные соединения обладают характерной окраской, что позволяет использовать их для обнаружения ионов в растворе. Так, катионы Fe2+ или Fe3+ можно определить по образованию темно-синих осадков «турнбулевой сини» Fe§+ [Fe3+(CN)6]2 и «берлинской лазури» Fe^+ [Fe2+(CN)6]3 при реакциях с феррицианид-ионом [Fe(CN)6]3
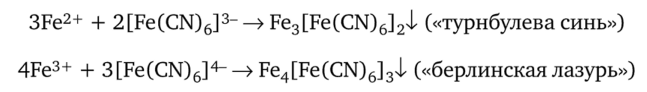
Тетратиоцианатный комплекс кобальта(П) не очень устойчив (lgK = 2,20, т. е. Ку = 1,6 • 102), и в разбавленных растворах равновесие сильно смещено влево. Ионы Fe3+ также образуют с тиоцианат-ионами окрашенные (в красный цвет) тиоцианатные комплексы [Fe(SCN)n]3-n:
При гс = 6 константа устойчивости комплекса железа(Ш) Ку — 1,7 х х 103 (IgKy = 3,23), т. е. этот комплекс несколько устойчивее тиоцианатного комплекса кобальта(Н). На фоне окрашенного комплекса железа(Ш) окраску соединения кобальта(Н) заметить практически невозможно, т. е. катионы Fe3+ мешают открытию ионов Со2+. Мешающее действие катионов железа(III) можно подавить введением в раствор фторид-ионов F-. В присутствии этих анионов железо (III) связывается в очень прочный бесцветный комплекс [FeF6]3-:
с константой устойчивости Ку = 7 • 1011 (lgKy = 11,86), на несколько порядков превышающей константу устойчивости тиоцианатного комплекса железа(Ш). Последний в присутствии фторид-ионов разрушается и переходит во фторидный комплекс железа(Ш):
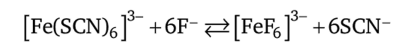
Концентрационная константа этого равновесия (Кс = 4,1 • 108) достаточно большая, поэтому равновесие практически полностью сдвинуто вправо — в сторону образования фторидного комплекса железа(Ш), т. е. тиоцианатный комплекс практически разрушается. Ион кобальта Со2+ в этих условиях не образует прочных фторидных комплексов.
Для маскирования ионов металлов (устранения их мешающего действия) используют их связывание в хлоридные, тиосульфатные, аммиачные комплексы, в комплексонаты. Основное условие, которое должно при этом выполняться, состоит в том, что образующийся комплекс, в который связываются мешающие ионы, должен обладать высокой устойчивостью (высоким значением константы устойчивости Ку), а определяемый ион, напротив, не должен образовывать устойчивые комплексы с маскирующим агентом.
Изменение окислительно-восстановительных потенциалов (ОВП) редокс-систем. Образование различных комплексов влияет на величину ОВП редокс-пар. Рассмотрим, например, редокс-пару pt4+|pt2+, которой соответствует полуреакция Pt4+ +2е