Наследственные болезни человека. "Наследственные болезни человека и возможности их лечения"
 Скачать 106.05 Kb. Скачать 106.05 Kb.
|

|
Глава III. классификация наследственных заболеваний человека В 90-х годах XX в. предложена рабочая классификация НБ человека, включающая: 1) синдромы, обусловленные хромосомными аномалиями (хромосомные болезни); 2) болезни, вызванные мутацией отдельного гена (генные или менделеевские болезни); 3) мультифакториальные заболевания (болезни с наследственным предрасположением); 4) болезни с нетрадиционным типом наследования, например, митохондриальные болезни, связанные с мутациями ДНК митохондрий; 5) генетические болезни соматических клеток (опухоли, старение, аутоиммунные болезни). III.1. Генные болезни iii.1.1. характеристика Менделевские болезни с позиции генетики стали изучаться уже в начале нашего века. В эту группу входят моногенно обусловленные патологические состояния, наследуемые в соответствии с законами Менделя. Большинство генных патологий обусловлено мутациями в структурных генах, осуществляющих свою функцию через синтез полипептидов — белков. Любая мутация гена ведет к изменению структуры или количества белка. Начало любой генной болезни связано с первичным эффектом мутантного аллеля. Основная схема генных болезней включает ряд звеньев: мутантный аллель → измененный первичный продукт → цепь биохимических процессов в клетке → органы → организм. В результате мутации гена на молекулярном уровне возможны следующие варианты: синтез аномального белка; выработка избыточного количества генного продукта; отсутствие выработки первичного продукта; выработка уменьшенного количества нормального первичного продукта. Не заканчиваясь на молекулярном уровне в первичных звеньях, патогенез генных болезней продолжается на клеточном уровне. При различных болезнях точкой приложения действия мутантного гена могут быть как отдельные структуры клетки — лизосомы, мембраны, митохондрии, пероксисомы, так и органы человека. III.1.2. Принципы классификации Существует два принципа классификации менделеевских болезней, в основе которых лежат следующие принципы: генетический в зависимости от типа наследования и в зависимости от функциональной значимости первичных продуктов соответствующих генов. Согласно первому все генные болезни можно разделить на: аутосомные (доминантные и рецессивные) X-сцепленные (доминантные и рецессивные) Y-сцепленные (голандрическое наследование) митохондриальные Второй принцип осуществляет подразделение таким образом: наследственные нарушения ферментных систем (энзимопатии) дефекты белков крови (гемоглобинопатии) дефекты структурных белков ( коллагеновые болезни) генные болезни с невыясненным биохимическим дефектом В основе энзимопатий лежат либо изменения активности фермента, либо снижение эффективности его синтеза. У гетерозигот-носителей мутантного гена присутствие нормальной аллели обеспечивает сохранение 50% активности фермента по сравнению с нормальным состоянием. Поэтому наследственные дефекты ферментов клинически проявляются у гомозигот, а у гетерозигот недостаточная активность фермента выявляется специальными исследованиями. В зависимости от характера нарушений обмена веществ в клетках среди энзимопатий различают следующие формы: Наследственные дефекты обмена углеводов (галактоземия – нарушение метаболизма молочного сахара, недостаточность фермента галактозо-1-фосфат-уридил-трансферазы; мукополисахаридозы – нарушение расщепления полисахаридов и другие). Наследственные дефекты обмена липидов и липопротеинов (сфинголипидозы – нарушение расщепления структурных липидов; нарушения обмена липидов плазмы крови, сопровождающиеся увеличением или уменьшением содержания в крови холестерина, лецитина). Наследственные дефекты обмена аминокислот (альбинизм – отсутствие кожных пигментов, седина, дефекты зрения, следствие недостаточности фермента – тиразиназы; фенилкетонурия – при наличии в пище фенилаланина развивается резко выраженное слабоумие, недостаточность фермента фенилаланингидроксилазы; тирозиноз - задержка нервно-психического развития, нарушение обмена тирозина; алкаптонурия - проявляется в среднем возрасте в виде деформирующих артритов конечностей и позвоночника) Наследственные дефекты обмена витаминов (гомоцистинурия – развивается как результат генетического дефекта кофермента витаминов B6 и B12). Наследственные дефекты обмена пуриновых и пиримидиновых азотистых оснований (синдром Леша-Найяна, связанный с недостаточностью фермента, который катализирует превращение свободных пуриновых оснований в нуклеотиды). Наследственные дефекты биосинтеза гормонов (адреногенитальный синдром, связанный с мутациями генов, которые контролируют синтез андрогенов; тестикулярная феминизация, при которой не образуются рецепторы андрогенов). Наследственные дефекты ферментов эритроцитов (некоторые гемолитические несфероцитарные анемии, характеризующиеся нормальной структурой гемоглобина, но нарушением ферментной системы, участвующей в анаэробном расщеплении глюкозы). Гемоглобинопатии - это группа наследственных заболеваний, вызываемых первичным дефектом пептидных цепей гемоглобина и связанным с нарушением его свойств и функций. Нарушения функций гемоглобина, возникающие в результате таких изменений структуры ά- и β-глобиновых генов, ведут к появлению заболеваний, которые можно разделить на 4 основные группы. Гемолитические анемии. Проявляются в распаде эритроцитов, зависящем от неустойчивости гемоглобина. Метгемоглобинемия. Обусловлены ускоренным окислением двухвалентного железа до трехвалентного и образованием гемоглобина М. Эритроцитоз. Заключается в образовании большего, чем обычно, количества эритроцитов, что обусловлено повышенным сродством гемоглобина к кислороду, который с трудом высвобождается в тканях. Серповидно-клеточная анемия. Заключается в замене гемоглобина HbA на HbS, который отличается растворимостью и кристаллизацией в условиях гипоксии, что приводит к изменению формы эритроцитов (см. I.1.). Заболевания первых трёх групп наследуются по доминантному типу, так что гетерозиготы по мутантному гену страдают нарушением здоровья. Наследование серповидно-клеточной анемии при обычных условиях осуществляется по рецессивному типу, но в условиях недостатка кислорода (высокогорье) гетерозиготы также страдают анемией. В основе возникновения коллагеновых болезней лежат дефекты биосинтеза и распада коллагена – структурного компонента соединительной ткани (болезнь Эллерса-Данлоса, болезнь Марфана). К наследственным болезням с невыясненным первичным биохимическим дефектом относится подавляющее большинство моногенных наследственных болезней. Среди них можно выделить: муковисцидозы - частота 1:2500, наследственное поражение экзокринных желёз и железистых клеток организма, выделение ими густого, измененного по составу секрета и связанные с этим последствия. ахондроплазия – частота 1:100000, заболевание костной системы, при котором наблюдаются аномалии развития хрящевой ткани преимущественно в эпифизах трубчатых костей и костях основания черепа. Миопатии (мышечные дистрофии) – заболевания, связанные с поражением поперечно-полосатых и гладких мышц (миопатия Дюшена). III.2. Хромосомные болезни и их характеристика Хромосомные болезни связаны с изменением числа или структуры хромосом. Характерным отличием большинства из них от болезней, вызываемых генными мутациями, является то, что они не передаются по наследству, а повторно возникают. Хромосомные и геномные мутации возникают при гаметогенезе родителей, а также непосредственно в зиготе на ранних стадиях дробления. У человека известны все типы хромосомных и геномных мутаций, включая полиплоидию. Однако такие полиплоиды либо рождаются мёртвыми, либо живут несколько дней. III.2.1 Количественные нарушения аутосом Синдром Дауна впервые был описан в 1866 году английским педиатром Л.Дауном, но только в 1959 году французским генетиком Дж. Леженом было доказано, что это заболевание хромосомной природы, результат трисомии по хромосоме 21. Частота этого заболевания 1:700-800 новорождённых. В подавляющем большинстве случаев обнаруживается простая трисомия 21 (кариотип -47ХХ (ХУ)+21). Около 4% случаев обусловлены транслокационной формой, и в 2% случаев развивается мозаицизм. Диагноз обычно не сложен и основывается на характерных сочетаниях морфологических, функциональных особенностей и результатов цитогенетического исследования. Минимальными диагностическими признаками являются: умственная отсталость, мышечная гипотония, плоское лицо, монголоидный разрез глазных щелей, трисомия 21. В отношении мозаичной формы существует мнение о том, что у пациентов наблюдается большой клинический полиморфизм, варьирующий от нормального фенотипа до полной клинической картины синдрома. Эти отличия объясняются числом трисомных клеток, однако прямой зависимости между процентов клеток с трисомией и степенью умственного развития нет. Хорошо известно, что дети с этим синдромом чаще рождаются у женщин старше 35 лет. Транслокационные формы, наоборот, встречаются у молодых родителей. Мозаичные формы встречаются с одинаковой частотой во всех возрастных группах. Зависимость частоты рождения детей с синдромом Дауна от возраста матери показана на графике: 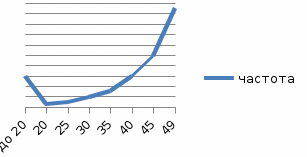 Согласно статистическим данным частота заболевания синдромом Дауна по Дзержинскому району города Ярославля составляет 1:2169, что в 3,1 раза меньше среднестатистической цифры (1:700). Объяснить этот факт можно тем, что: 1) В городе работает центр планирования семьи и репродукции «Брак и семья», в котором проводится медико-генетическое консультирование для выявления наследственной патологии (см. глава V); 2) Снижение производственной деятельности людей (закрытие промышленных предприятий, сокращение рабочих) и возрастание роли непроизводственной сферы (сфера услуг, транспорт, туризм, торговля), в пределах которой число мутагенов, действующих на организм, меньше. 3) Пропаганда медико-генетических знаний среди врачей и населения. Генетическая природа синдрома Патау, трисомия 13, была расшифрована в 1960г. американским генетиком Патау. Частота данного заболевания составляет 1 на 6000 рождений, занимая второе место по частоте встречаемости (после синдрома Дауна) среди полных аутосомных трисомий. Простая полная трисомия 13 (47, ХХ (ХУ) + 13) как следствие нерасхождения этой пары хромосом в мейозе у одного из родителей (обычно у матери) встречается в 85% случаев, остальные обусловлены транслокациями. Случаи мозаицизма крайне редки. Фенотипические признаки синдрома настолько характерны, что позволяют практически сразу заподозрить это заболевание. Особенно обращают на себя внимание аномалии черепа и лица – микроцефалия, скошенный лоб, узкие глазные щели, запавшее переносье, низкорасположенные и деформированные ушные раковины. Наиболее характерными внешними пороками развития являются расщелина губы и полидактилия. Врожденные пороки сердца встречаются у 80% детей, почек – 60%, органов зрения – 70%, ЦНС – 100% случаев. У девочек наблюдаются удвоение матки влагалища, у мальчиков - гипоплазия полового члена. Продолжительность жизни у детей с синдромом Патау резко снижена, на первом году погибают 95%. Все дети имеют тяжёлую умственную отсталость (глубокая идиотия). Синдром Эдвардса получил название по имени английского цитогенетика, впервые описавшего его хромосомную природу в 1960 году при обследовании ребенка с множественными пороками и аномалиями развития. Оказалось, что причиной синдрома была полная трисомия по 18 хромосоме, возникающая в результате нерасхождения 18 хромосомы в время мейоза. Частота синдрома 1:7000. Характерно сочетание специфических клинических проявлений: долихоцефалия, гипоплазия нижней челюсти и микростомия, узкие и короткие глазные щели, маленькие низко расположенные ушные раковины, характерное сгибательное положение пальцев кисти, выступающий затылок. При синдроме практически постоянны пороки сердца, ЖКТ, почек, половых органов. Продолжительность жизни больных резко снижена. На первом году погибают 90%, к 3-м годам – более 95%. Причиной смерти являются пороки сердечно-сосудистой системы, кишечника, половых органов. Все выжившие имеют глубокую степень олигофрении (идиотию). III.2.2. Количественные нарушения половых хромосом Изменения числа половых хромосом могут возникать в результате нарушения расхождения как в первом, так и во втором делении мейоза. Нарушение расхождения в первом делении приводит к образованию аномальных гамет: у женщин – ХХ и 0 ( в последнем случае яйцеклетка не содержит половых хромосом); у мужчин – ХУ и 0. При слиянии гамет во время оплодотворения возникают количественные нарушения половых хромосом. Основные признаки аномалий половых хромосом
Реже среди аномалий половых хромосом встречаются тетрасомии Х и синдром дисомии по Y-хромосоме. При первом отмечается высокий рост, телосложение по мужскому типу, эпикант, уплощенное переносье, высокое нёбо, деформированные и аномально расположенные ушные раковины, поперечная ладонная складка. У женщин с данным заболеванием описаны различные нарушения менструального цикла, бесплодие, преждевременный климакс. Снижение интеллекта отмечено у 2/3 больных. В случае дисомии по Y-хромосоме (47, XXY) у большинства больных отмечается ускорение темпов роста в детском возрасте, высокий рост, грубые черты лица, надбровные дуги, увеличенная нижняя челюсть, большие ушные раковины, высокое нёбо, аномальный рост зубов с дефектами зубной эмали, деформация коленных и локтевых суставов. Интеллект негрубо снижен или в норме. Характерны эмоционально – волевые нарушения: агрессивность, взрывчатость, импульсивность. Продолжительность жизни не отличается от среднепопуляционной. III.2.3 Структурные нарушения аутосом Хромосомные болезни, вызванные хромосомными мутациями, очень многочисленны. Клинически и цитогенетически идентифицировано более 100 синдромов. Один из них синдром «кошачьего» крика. Частота его среди новорожденных составляет 1:45000. Причиной заболевания является делеция части короткого плеча 5-й хромосомы (5р-). Наиболее характерным симптомом этого заболевания является специфический плач новорожденных, похожий на кошачий крик, возникновение которого связано с изменениями гортани – сужением, мягкостью хрящей, отечностью, уменьшением надгортанника. У этих детей выявляются микроцефалия, низкорасположенные и деформированные ушные раковины, лунообразное лицо, эпикант, монголоидный разрез глаз, мышечная гипотония. Дети резко отстают в физическом и умственном развитии. Врожденные пороки развития внутренних органов встречаются редко, наиболее часто поражается сердце. Все больные имеют тяжёлую степень умственной отсталости. Продолжительность жизни с синдром 5р- значительно выше, чем у пациентов с аутосомными трисомиями. III.3. Мультифакториальные заболевания Возникновение широко распространённых заболеваний, которые вносят наибольший вклад в заболеваемость, инвалидизацию и смертность населения, обусловлено взаимодействием наследственных факторов и разнообразных факторов внешней среды. Эту многообразную группу патологий называют болезнями с наследственным предрасположением или мультифакториальной патологией. Среди них имеются моногенные, вызванные одним патологически изменённым геном, и полигенные, определяемые многими генами, которые в нормальном состоянии, но при определённом воздействии среды создают предрасположение к появлению заболевания. Первая группа включает патологические реакции на действие различных факторов, например, лекарственных средств, пищевых добавок, физических и биологических агентов. Вторая группа более многочисленна. В неё входят такие заболевания, как псориаз, сахарный диабет, шизофрения, ревматоидный артрит. Например, люди с II(A) группой крови чаще заболевают раком желудка, матки, молочной железы, ревматизмом. III.4. Болезни с нетрадиционным типом наследования В последние годы стало очевидно, что далеко не все случаи наследственной патологии у человека можно рассматривать как результат менделирующих генных мутаций, хромосомных аномалий или как МФЗ. В настоящее время описано достаточно много заболеваний, которые в современной классификации наследственной патологии человека объединяют в отдельную группу: болезни с нетрадиционным типом наследования. Среди них различают: болезни импринтинга, митохондриальные болезни, болезни экспансии тринуклеотидных повторов с явлением антиципации и др. III.5. Генетические болезни соматических клеток Генетические болезни соматических клеток выделены в отдельную группу наследственной патологии недавно. Поводом к этому послужило обнаружение при злокачественных новообразованиях специфических хромосомных перестроек в клетках, вызывающих активацию онкогенов (ретинобластома, опухоль Вильмса). Эти изменения в генетическом материале клеток являются этиопатогенетическими для злокачественного роста и поэтому могут быть отнесены к категории генетической патологии. Уже имеются первые доказательства того, что спорадические случаи врождённых пороков развития являются результатом мутаций в соматических клетках в критическом периоде эмбриогенеза. Следовательно, такие случаи можно рассматривать как генетическую болезнь соматических клеток. Весьма вероятно, что аутоиммунные процессы и старение могут быть отнесены к этой же категории генетической патологии. |