Клиническая химия в диагностике и лечении. Обмен натрия и воды обмен калия лечение диуретиками гомеостаз ионов водорода
 Скачать 3.99 Mb. Скачать 3.99 Mb.
|

ВРОЖДЕННЫЕ НАРУШЕНИЯ ОБМЕНА ВЕЩЕСТВБиохимические особенности организма определяют приблизительно 50 000 пар генов, передаваемых из поколения в поколение через хромосомы. Индивидуальные вариации возникают в результате случайного отбора и рекомбинации во время редукционного деления (мейоза), а также в результате периодических мутаций. Последствия таких вариаций бывают различными: от изменении, несовместимых с жизнью, до возникновения биохимических особенностей, которые, если и удается обнаружить, то лишь с помощью специальных методов исследования. К этой последней категории относятся генетические вариации таких белков плазмы крови, как трансферрины и гаптоглобины, изменения которых важны в связи с популяционными и генетическими исследованиями, но не всегда приводят к функциональным расстройствам. Наряду с двумя указанными выше крайними вариантами существует множество промежуточных вариаций, приводящих к развитию функциональных аномалий, для обозначения которых и применяют термин врожденные нарушения обмена веществ. ОБЩИЕ ПРИНЦИПЫ Поскольку последовательность оснований в нитях ДНК, представляющих собой гены, кодирует через РНК структуру белков, не удивительно, что большинство (если не все) биохимических аномалий можно объяснить нарушениями биосинтеза одного пептида. Такие аномалии могут быть обусловлены наличием либо измененного структурного гена, кодирующего образование вариантного белка, либо измененного регуляторного гена, что приводит к нарушению функционирования одного или нескольких структурных генов и, следовательно, к вариациям содержания одного или нескольких белков с неизмененной структурой. В большинстве примеров, которые мы рассматриваем в этой главе, измененные белки являются ферментами. Возможные последствия для обмена веществ Последствия недостаточности одного фермента в цепи реакций обмена веществ могут проявляться поразному. Предположим, что превращение соединения А в соединение Б катализирует фермент Е и что соединение В встречается на альтернативном пути превращений (рис. 41). Последствиями недостаточности Е могут быть следующие явления: 1) недостаточность продукта ферментативной реакции (Б). В качестве примеров укажем на недостаточность кортизола при врожденной гиперплазии надпочечников и на гипогликемию при некоторых формах гликогенозов; Рис. 41. Схема альтернативных путей биохимических превращений. 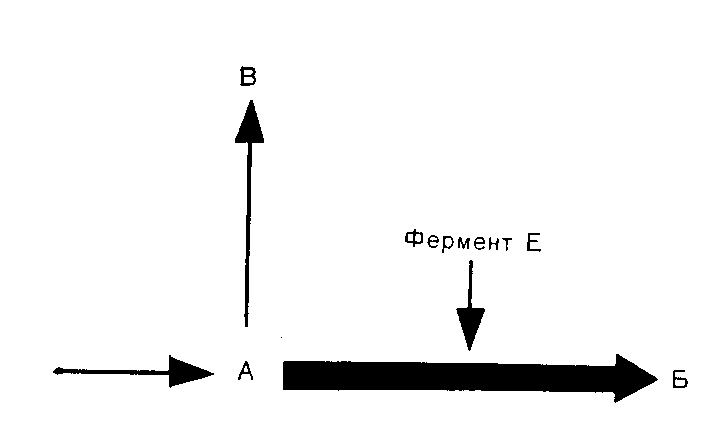 2) накопление вещества, превращение которого катализирует фермент (А) (например, фепилаланин при фенилкетонурии). При многих лизосомных болезнях накопления, вещества, в норме подвергающиеся гидролизу в лизосомах, накапливаются в них в связи с недостаточностью одного из ферментов; 3) отклонение на альтернативный путь с образованием некоторых биологически активных соединений (В). К этой группе явлений относится вирилизация, обусловленная андрогенами, при врожденной гиперплазии надпочечников. Если метаболическое превращение в целом регулируется по принципу обратной связи конечным продуктом, то эффекты двух последних типов аномалий будут особенно значительными. Так, например, при врожденной гиперплазии надпочечников недостаточность кортизола стимулирует синтез стероидов и, следовательно, накопление андрогенов, в результате чего развивающаяся вирилизация еще более усиливается. Факторы внешней среды могут модифицировать (или даже полностью определять) клинические проявления некоторых врожденных нарушений обмена веществ. Так, например, поскольку у женщин во время менструации и беременности происходит потеря организмом железа, при идиопатическом гемохроматозе у женщин накапливается меньше железа, чем у мужчин при этом же заболевании. У больных с вариантными формами холинэстеразы длительный паралич развивается только после введения миорелаксанта дитилина (с. 386); у некоторых больных с недотаточностью глюкозо6фосфатдегидрогеназы гемолиз начинается только после приема внутрь таких лекарственных средств, как примахин. Такие больные представляются «здоровыми» в отсутствие контактов с современными лекарственными средствами. Когда возникает предположение о наличии врожденного нарушения метаболизма Возможность врожденного нарушения обмена веществ следует предполагать, если в раннем или позднем детском возрасте обнаруживают клинические или биохимические аномалии (особенно у двух пли большего числа детей в одной семье). Следующие явления заслуживают особого внимания в этой связи (если для их возникновения нет очевидных причин): 1) нарушение развития; 2) рвоты; 3) гипогликемия; 4)' особый запах или окраска пеленок; 5) гепатоспленомегалия; 6) желтуха; 7) задержка умственного развития, припадки, спастическое состояние мышц; 8) метаболический ацидоз; 9) почечные камни; 10) рахит, не поддающийся лечению. Клиническое значение врожденных нарушений метаболизма Выявление многих врожденных нарушений обмена веществ представляет только академический интерес, так как клинически они не проявляются. В тех случаях, когда не разработаны эффективные методы лечения, может оказаться важной диагностика как основа проведения генетического консультирования. Распознавание ряда заболевании в рапном детским возрасте жизненно важно, поскольку лечение может предотвратить развитие необратимых клинических явлений и гибель больного. Среди наиболее важных заболеваний такого типа назовем фенилкетонурию, галактоземию, болезнь «моча с запахом кленового сиропа». При ряде заболеваний показаны обследования родственников больного как для предупреждения дальнейшего развития заболевания, так и в связи с необходимостью избегать воздействия усугубляющих патологическое состояние факторов. Примерами патологических состояний такого типа являются аномалии холинэстеразы, недостаточность глюкозо6фосфатдегидрогеназы, острые порфирии, гемохроматоз, цистинурия, болезнь Уилсона. Примерами патологических состояний, поддающихся симптоматическому лечению, являются врожденный нефрогенньш несахарный диабет, врожденная недостаточность дисахаридазы, болезнь Хартнапа. Некоторые врожденные нарушения обмена веществ полностью (или почти полностью) безвредны. Они важны в том смысле, что могут приводить к диагностическим ошибкам или напрасно беспокоить пациента. Примерами таких состояний являются ренальная гликозурия, алкаптонурия, болезнь Жильбера. Наконец, некоторые врожденные нарушения обмена веществ могут клинически проявиться только после достижения половой зрелости. В этих случаях желательна генетическая консультация кровных родственников больного. Примером заболевания такого типа может быть болезнь Уилсона. Лабораторная диагностика врожденных нарушений метаболизма О недостаточности фермента обычно судят косвенно по повышению концентрации исходного вещества, которое в норме подвергается биохимическим превращениям, катализируемым данным ферментом (например, фенилаланин при фенилкетонурии). Прямые определения активности таких ферментов проводят только в специализированных центрах, но по возможности диагноз во всех случаях следует подтверждать этим методом. Пренатальная диагностика некоторых врожденных нарушений метаболизма возможна путем исследований клеток амниотической жидкости, полученных на ранних стадиях беременности и клуьтивируемых in vitro. Скрининг для выявления врожденных нарушений метаболизма Учитывая важность раннего проведения лечения, многие страны утвердили программы скрининга всех новорожденных для выявления врожденных нарушений метаболизма, особенно фенилкетонурии. Для тестирования можно использовать кровь (полученную после прокола кожи на пятке) или мочу. Кровь (или в более редких случаях мочу) часто собирают на фильтровальную бумагу; в таком виде ее легко доставлять в лабораторию. Важно учитывать время отбора проб для анализов, чтобы избежать получения ложноотрицательных результатов. Соединения, лежащие на путях метаболизма выше точки блокировки ферментативных превращений (например, фенилаланин при фенилкетонурии или галактоза при галактоземии), накапливаются только после того, как ребенок начинает получать с пищей соответствующие предшественники (такие как белковые или молочные продукты). При скрининговом обследовании детей, которых считают здоровыми, кровь обычно берут на 6—9й день жизни. Аномальные метаболиты могут быть не определены в моче до 4—6 нед после рождения, если почечный порог для них относительно высок. Полученный при скрининге положительный результат следует подтвердить путем количественного анализа или повторного тестирования. Многие обнаруживаемые аномалии являются транзиторными. В конце этой главы мы приводим ссылку на статью, в которой обсуждаются типичные ошибки, допускаемые при интерпретации результатов скринингового обследования новорожденных с целью выявления врожденных нарушений метаболизма, и указывается на ответственность исследователей, проводящих такую работу. Лечение при врожденных нарушениях метаболизма. Некоторые врожденные нарушения метаболизма поддаются лечению путем доставки в организм недостающего метаболита или путем ограничения поступления в пищеварительный тракт предшественников нарушенных процессов обмена веществ. Иногда могут быть удалены накапливающиеся продукты (например, железо при гемохроматозе). Характер наследования Следующий раздел представляет собой только краткий обзор; подробное изложение вопроса имеется в книгах по генетике. Любой наследуемый признак определяет пара генов на гомологичных хромосомах (по одной от каждого из родителей). Аллелями называют различные гены, определяющие один и тот же признак. Индивидуум, обладающий двумя идентичными аллелями является гомозиготным в отношении данного гена или наследуемого признака; если он имеет два различных аллеля, то он гетерозиготный. Носителями генов могут быть половые хромосомы (х и у) или аутосомы (сходные у представителей обоих полов); при этом характер наследования различен. Аутосомное наследование 1. Предположим, что один из родителей (родитель 1 в приведенном ниже примере) является носителем аномального гена (А). Если Н обозначает нормальный ген, то возможности комбинаций генов у потомства показаны в квадрате. 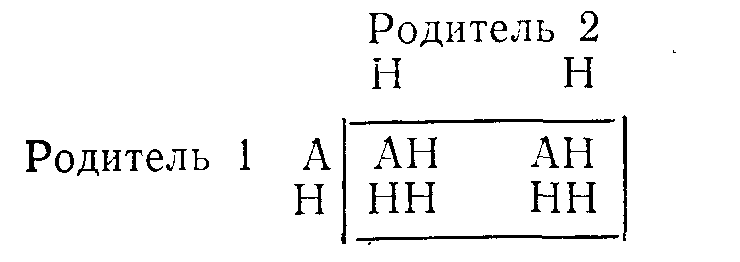 Видно, что на основе данных статистики половину потомков следует считать носителями одного аномального гена (АН); они будут гетерозиготными в отношении этого гена, как и родитель 1. Ни один из потомков не будет гомозиготным в отношении аномального гена (АА). 2. Если оба родителя гетерозиготны, то четверть их потомков (при общем большом числе) будет гомозиготна (АА) и половина гетерозиготна. 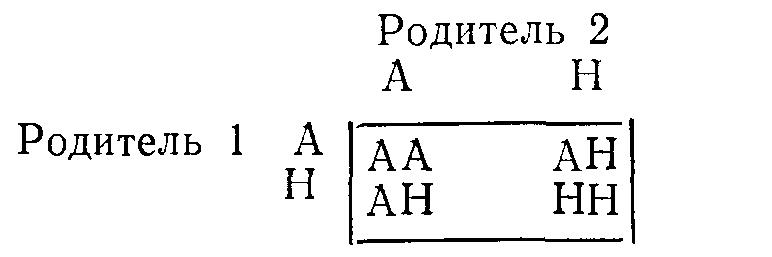 3. Если один родитель гомозиготен, а другой нормален, то все потомки будут гетерозиготными. Поскольку гены, определяющие клинически диагностируемые аномалии, как правило, встречаются редко, приведенный выше пример 1 статистически наиболее вероятен. При браках между родственниками пример 2 становится более вероятным, так как среди кровных родственников более вероятна возможность наличия носителей одинаковых аномальных генов, чем среди людей, не являющихся родственниками. Последствия носительства аномального гена зависят от его относительной мощности по сравнению с нормальным. Доминантный ген вызывает аномалии как у гетерозиготных, так и гомозиготных носителей, хотя степень поражения может быть более высокой у гомозигот. Так, в примере 1 будут поражены родитель 1 и половина потомков, а в примере 2 — оба родителя и 75% потомков. Характерно проявление аномалий в последующих поколениях. Рецессивный ген вызывает аномалии только у гомозиготных индивидуумов. Так, в примере 1 ни родители, ни потомки не будут поражены, а в примере 2 родители будут казаться здоровыми, но 25% потомков будут поражены. Характерно проявление одного или нескольких случаев заболевания лишь в одном поколении (при клинически здоровых родителях). Понятия доминантный и рециссивный относительны. Доминантный ген может не проявлять себя (неполная пенетрантность) и таким образом как бы «пропускать» поколение. Различной может быть степень экспрессии гена и, следовательно, степень выраженности аномалии, которую он определяет. Наконец, рецессивный ген, вызывающий заболевание только у гомозигот, может быть тем не Meiiee обнаружеи с помощью биохимических тестов п у гетерозиготных носителей. Наследование, сцепленное с полом Для некоторых аномальных генов носителями являются только половые хромосомы, почти всегда Ххромосомы. Х-сцепленное рецессивное наследование. Женщины являются носителями двух Ххромосом, а мужчины — одной Х и одной Y. При Хсцепленном рецессивном наследовании аномальная Ххромосома (Ха) находится в латентном состоянии, если она сочетается с нормальной Ххромосомой, но становится активной при сочетании с Yхромосомой. Если мать является носительницей Ха, она будет клинически здоровой, но половина (статистически) ее сыновей будут поражены (YXa). Половина дочерей будут носительницами (ХХа), но клинически все дочери будут здоровыми. 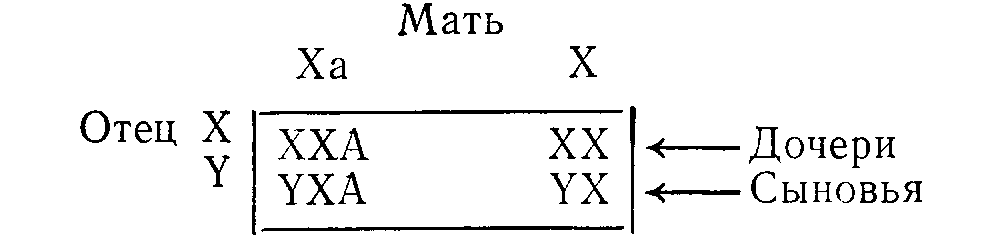 Если отец поражен заболеванием и мать является носительницей двух нормальных генов, ни один из сыновей не будет поражен, но все дочери будут гетерозиготными носительницами. 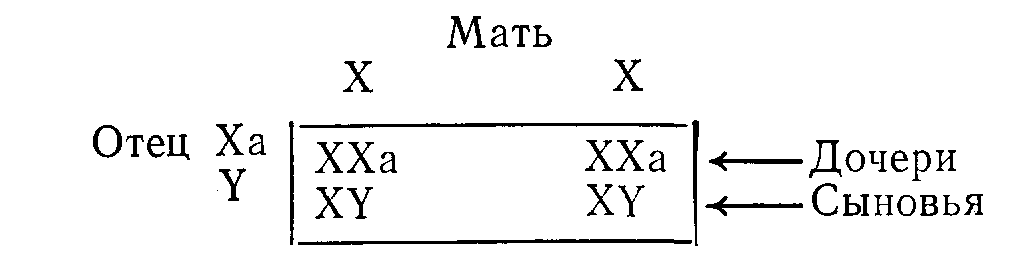 Для Х-сцепленного рецессивного наследования характерно проявление врожденных заболеваний только у потомков мужского пола, тогда как гетерозиготными носительницами являются женщины. Клинические симптомы заболевания у женщин встречаются редко, только когда они гомозиготны по аномальному гену. Такая ситуация может возникать в тех случаях, если поражен отец, а мать является гетерозиготной носительницей. Классическим примером Хсцепленного рецессивного наследования является гемофилия. Х-сцепленное доминантное наследование. При этом типе наследования поражены как ХХа, так и YXa (женщины и мужчины), например, при врожденной гипофосфатемии. Множественные аллели. В некоторых случаях один и тот же признак могут определять несколько аллелей. Разные сочетания пар могут при этом приводить либо к развитию различных заболеваний (например, некоторых гемоглобинопатий), либо к возникновению вариации, которые удается обнаружить только при биохимическом тестировании (например, вариации белков плазмы крови). ЗАБОЛЕВАНИЯ, ОБУСЛОВЛЕННЫЕ ВРОЖДЕННЫМИ НАРУШЕНИЯМИ МЕТАБОЛИЗМА Мы рассмотрим лишь очень немногие из числа изученных врожденных нарушений метаболизма. Избранные нами примеры иллюстрируют некоторые принципы, упомянутые выше. Сделанный выбор, безусловно, определяется нашими представлениями об относительной важности отдельных вопросов. С этими представлениями, возможно согласятся не все исследователи. На с. 391 были перечислены некоторые аномалии, представляющиеся наиболее важными для клиники. В более полном (но, конечно, не исчерпывающем) перечне, представленном на с. 406, врожденные патологические состояния систематизированы; по возможности указав характер их наследования. Многие из этих заболеваний мы уже кратко упоминали в соответствующих главах этой книги. Остается рассмотреть лишь немногие патологические состояния, которые, по нашему мнению, представляются относительно более важными по сравнению с другими, не упоминаемыми в тексте. Частота заболеваний. Все врожденные нарушения метаболизма встречаются очень редко. В ряде стран в ходе осуществления программ скрининга была приблизительно оценена частота заболеваний, связанных с врожденными нарушениями метаболизма, Из числа заболеваний, рассматриваемых ниже, фонплкетонурия, болезнь Хартнапа, цистинурия, врожденная иминоглицинурия в гистидинемия распространены особенно широко (1 на 10000— 20 000); болезнь «моча с запахом кленового сиропа» встречается значительно реже (приблизительно 1 на 350 000). Аминоацидурия Поскольку нарушения метаболизма или экскреции аминокислот встречаются при многих врожденных аномалиях обмена веществ, аминоацидурия представляет собой один из первых симптомов, который следует искать в таких случаях. В норме аминокислоты фильтруются в почечных клубочках и поступают в проксимальные отдельг канальцев с концентрацией, равной таковой в плазме; почти все они здесь активно реабсорбируются. Таким образом, возможна аминоацидурия двух типов: 1) аминоацидурия перегрузки, когда в связи с повышением содержания аминокислот в плазме крови они поступают в проксимальный отдел канальцев в концентрациях, превосходящих реабсорбционную мощность клеток почечных канальцев; 2) почечная аминоацидурия, при которой содержание аминокислот в плазме крови снижено в результате потерь их из организма с мочой вследствие недостаточности реабсорбции в канальцах. На основании "данных о характере экскретируемых с мочой аминокислот можно различать два типа аминоацидурий: 1) специфическую аминоацидурию, когда чрезмерно экскретируется либо одна аминокислота, либо группа родственных аминокислот. Такая аминоацидурия, обусловленная как перегрузкой, так и почечной недостаточностью, почти всегда генетически предопределена; 2) неспецифическую амипоацидурию, когда происходит чрезмерная экскреция целого ряда аминокислот неродственных между собой. Такая аминоацидурия почти всегда является не врожденной, а приобретенной. Она может быть обусловлена перегрузкой как, например, при тяжелых заболеваниях печени, когда нарушение дезаминирования аминокислот приводит к повышению их содержания в плазме крови. Чаще встречается почечная аминоацидурия, связанная с неспецифическим поражением проксимальных отделов канальцев. В таких случаях, обозначаемых термином синдром Фанкони, другие вещества, реабсорбированные в проксимальных отделах канальцев, также выводятся из организма с мочой в чрезмерных количествах (фосфоглюкоаминоацидурия). При врожденных нарушениях метаболизма развитие синдрома Фанкони значительно чаще обусловлено вторичным повреждением почечных канальцев веществами, не подвергающимися нормальным биохимическим превращениям (как, например, медь при болезни Вильсона), чем первичным генетическим дефектом. Врожденные аномалии механизмов транспорта веществ Группы химически родственных аминокислот часто реабсорбируются в почечных канальцах при участии одних и тех же или взаимосвязанных механизмов. В некоторых случаях аналогичные группоспецифические механизмы участвуют в процессах всасывания в кишечнике, и при явлениях недостаточности бывают поражены как почечные канальцы, так и слизистая оболочка кишечника. Идентифицированы врожденные нарушения следующих группоспецифических механизмов транспорта аминокислот: 1) двухосновных (т. е. имеющих 2 аминогруппы в молекуле) аминокислот цистина, орнитина, аргинина и лизина (ЦОАЛ — полезный мнемонический прием) (цистинурия); 2) многих нейтральных аминокислот (имеющих одну амино и одну карбоксильную группу в молекуле) (болезнь Харнапа); 3) иминокислот, пролина и оксипролина, возможно, в сочетании с глицином (врожденная иминоглицинурия). Цистинурия Цистинурия обусловлена врожденной аномалией реабсорбции в почечных канальцах двухосновных аминокислот цистина, орнитина, аргинина и лизина, что приводит к чрезмерной экскреции с мочой этих 4 аминокислот. Аналогичное нарушение процессов транспорта аминокислот возможно и в слизистой оболочке кишечника, но, хотя всасывание цистина и понижается, но синтезируется в организме и его дефицит не развивается. Недостаточность реабсорбции в почечных канальцах приводит к высокой экскреции цистина с мочой. Поскольку его расгиордмосгь итносытельно невелика, он может выпадать в осадок в мочевых путях, что способствует образованию камней со всеми последующими осложнениями. Растворимость цистина такова, что только у гомозигот его концентрации в моче достигают уровня, при котором возможны выпадение цистина в осадок, кристаллурия и образование камней, хотя повышенную экскрецию цистина с мочой можно определить и у гетерозиготных носителей. Диагностика цистипурии основана на обнаружении чрезмерной экскреции с мочой цистипа и других характерных аминокислот. Данные о последних необходимы для того, чтобы дифференцировать гомозигот, в организме которых происходит образование камней, от цистинлизинурий у гетерозигот, а также от цистинурии, составляющей часть генерализованной аминоацидурий. Лечение при цистинурии направлено на предотвращение образования камней путем введения в организм днем и ночью больших объемов жидкости, что приводит к снижению концентрации цистина в моче. Растворимость цистина повышается также при подщелачивании мочи. Если эти меры оказываются неадекватными, можно попытаться вводить Dпеницилламин, в присутствии которого образуется более растворимое соединение. Различные генетические формы цистинурии наследуются по аутосомнорецессивному типу. Многие случаи бессимптомньге. Описанные выше относительно безвредные явления необходимо отличать от цистиноза. Это весьма редкое врожденное нарушение обмена цистина характеризуется его накоплением в клетках многих тканей. В почках такое нарушение обмена приводит к поражению канальцев и затем к развитию синдрома Фанкони. Эта аминоацидурия является неспецифической и имеет почечное происхождение. Больные умирают в раннем детском возрасте. Болезнь Хартнапа Болезнь Хартнапа, названная по фамилии больного, у которого она бьша впервые идентифицирована, представляет собой редкое, во интересное патологическое состояние, характеризуемое нарушением транспорта нейтральных аминокислот в почках и пищеварительном тракте. Большинство (если не все) клинических симптомов этой болезни может быть связано с уменьшением всасывания в кишечнике триптофана и с увеличением его экскреции с мочой. В норме триптофан частично превращается в никотинамид. Этот процесс имеет особенно важное значение, если поступление в организм никотинамида с продуктами питания ограничено. На этом примере можно видеть, как изменения условий внешней среды воздействуют на проявления врожденных нарушений обмена веществ. Клинические проявления болезни Хартнапа имеют перемежающий характер и напоминают симптомы пеллагры, а именно: 1) красная чешуйчатая сыпь на открытых поверхностях кожи; 2) обратимая мозжечковая атаксия: 3) спутанность сознания, выраженная в различной степени. То, что эти клинические симптомы обусловлены недостаточностью никотинамида, подтверждается данными о терапевтической эффективности введения данного витамина и наличием периода алиментарной недостаточности, часто предшествующего развитию проявлений заболевания. Несмотря на общую недостаточность процесса всасывания аминокислот, признаки обусловленных алиментарными факторами нарушений обмена белков отсутствуют, что, по-видимому, объясняется всасыванием интактных пептидов (очевидно, посредством других путей). Сопутствующим результатом лабораторных исследовании является экскреция с мочой чрезмерных количеств соединений индола. Эти соединения образуются в кишечнике при действии бактерий на неабсорбированный триптофан. Болезнь Хартнапа наследуется по рецессивному типу. Диагностика основана на обнаружении в моче характерного сочетания экскретируемых аминокислот. Существующие методы исследования не позволяют легко установить гетерозиготных носителей данного заболевания. Наследственная иминоглицинурия. Аномалия механизма трансапорта аминокислот приводит к повышению экскреции с мочой пролина, оксипролина и глицина, по их содержание в плазме крови остается в пределах нормы. Это явление представляется безвредным, но его необходимо отличать от других, более серьезных. случаев иминоглицинурии. Данное состояние наследуется по аутосомнорецессивному типу. Нарушения обмена аминокислот Описано множество врожденных нарушений обмена аминокислот. Большинство из них характеризуется повышенным содержанием соответствующих аминокислот в крови и аминоацидурией, обусловленной перегрузкой. Мы описываем в этом разделе.' лишь немногие, более подробно изученные, нарушения. Нарушения обмена ароматических аминокислот На рис. 42 представлена схема основных химических превращений этих аминокислот, а также известных в настоящее время нарушений активности ферментов, катализирующих эти превращения. Показано, что тирозин, который в норме образуется в организме из фенилаланина, является предшественником целого ряда важных соединений. Фенилкетонурия. Данное патологическое состояние вызывается аномалией системы фенилалапингидроксилазы, обычно самого фермента, но в некоторых случаях может быть нарушен биосинтез его кофактора тетрагидробиотерина. Таким образом, весьма сходные между собой аномалии биохимических процессов и клинические симптомы могут быть обусловлены несколькими различными врожденными нарушениями метаболизма. Поскольку фенилалашшгидроксилаза катализирует иревращепие енилаланина в тирозин, при рассматриваемом патологическом состоянии в крови накапливается феналаланин, который (в сочетании с такими продуктами его побочного превращения, как фенилпироиноградная кислота) экскретируется с мочой. Этот факт (экскреция фенилкетонов с мочой) получил отражение в названии заболевание широко практикуются скрининговые исследования новорожденных с целью раннего выявления случаев фенилкетонурии. Проблемы, связанные с этими обследованиями, мы обсудим ниже. 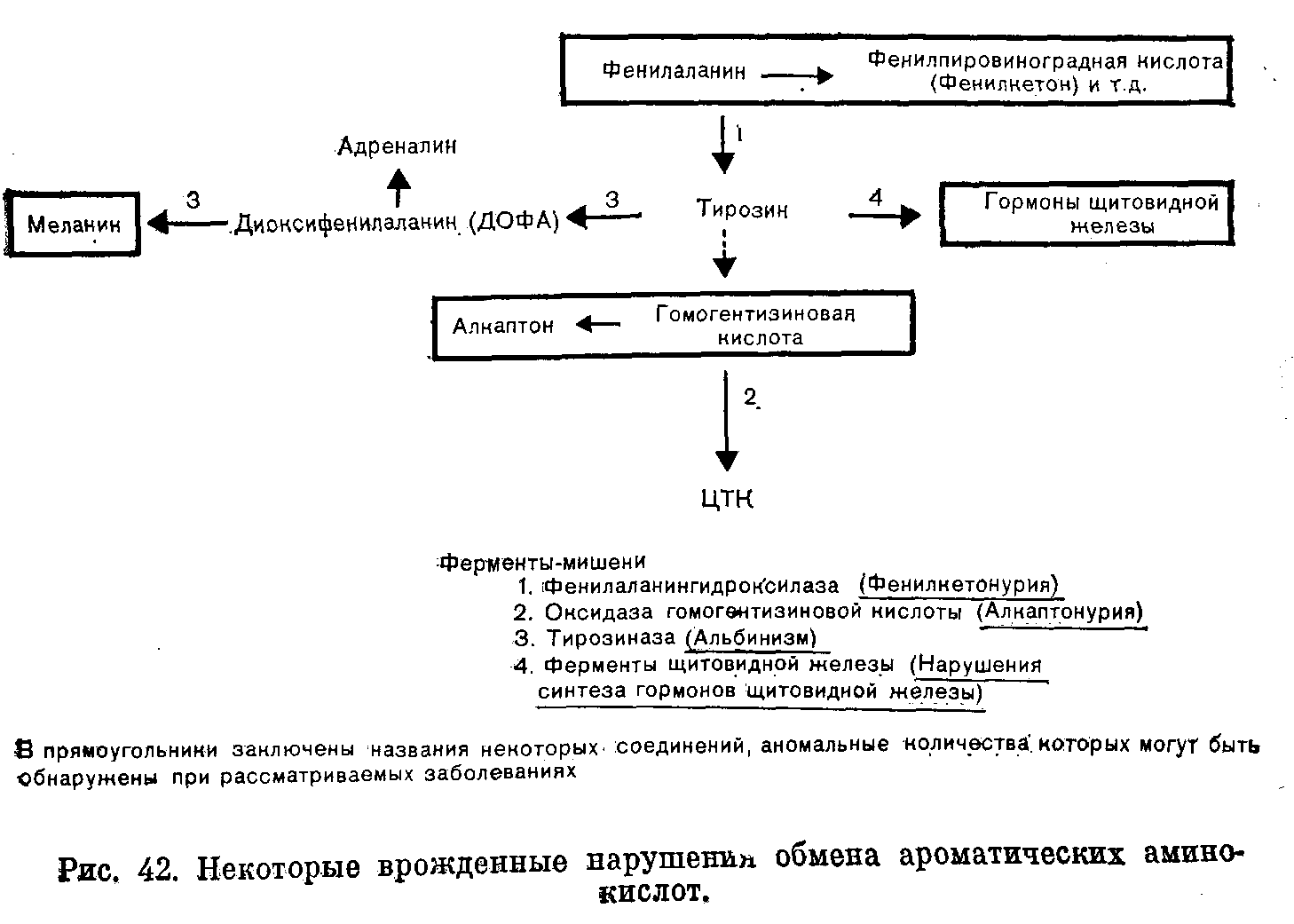 Отметим следующие клинические симптомы этой болезни: 1) раздражительность, затруднения при кормлении новорожденных, рвоты и припадки в течение первых нескольких недель жизни; 2) задержка психического развития в период между 4 и 6 мес жизни на фоне необычной психомоторной раздражительности 3) генерализованная экзема (во многих случаях); 4) тенденция к понижению меланинообразования. У многих больных бледная кожа, светлые волосы, голубые глаза. Диагностика. Концентрация фенилаланина может быть измерена в крови, взятой из прокола кожи на пятке. Эта методика пригодна для массовых скрининговых обследований. Лучше всего проверить тестирование в специализированных центрах. Особенно важное значение имеет время проведения теста. Рекомендуется проводить тестирование между 6 и 10 днями жизни (т. е. наканупс выписки ребенка из стационара). Фенилпировиноградная кислота мочи реагирует с хлорным железом или препаратом фенистикс. Однако такое тестирование может дать положительный результат только примерно через 6 нед; после рождения, когда уже могут развиться необратимые поражения головного мозга. После внедрения скрининговых тестов в клиническую практику стала очевидной возможность повышенного содержания фенилаланина в крови не только при фенилкетонурии, по и при других состояниях, в частности у недоношенных детей, что, по-видимому, обусловлено замедлением созревания ферментных систем. Такие случаи можно идентифицировать путем повторных тестирований крови и определения в крови содержания тирозина, концентрация которого не повышена при фенилкетонурии, но повышена при многих других состояниях. Описана нетипичная стойкая (персистирующая) гиперфенилаланинемия, не сопровождающаяся задержкой умственного развития. У детей, которые in utero подвергались воздействию высоких концентраций фенилаланина, если у их матерей была недиагностированная или нелеченая фенилкетонурия, могут быть признаки умственной отсталости и других врожденных аномалий, хотя сами по себе они не страдают фенилкетонурией (материнская фенилкетонурия). Лечение. Цель лечения — понижение уровней фенилаланина в крови с помощью диеты, бедной фенилаланином. Такое лечение является трудным, дорогим и утомительным для больных и их родителей; оно требует тщательного биохимического мониторинга концентрации фенилаланина в крови. Недостаточность фенилаланина сама по себе оказывает вредное воздействие. В состав диеты необходимо включать тирозин, являющийся предшественником. многих важных продуктов обмена (см. рис. 42). Фенилкетонурия наследуется по аутосомнорециссивному типу. Гетерозиготные носители клинически здоровы, но могут быть идентифицированы путем биохимического тестирования. Алкаптонурия. Врожденная недостаточность окспдазы гомогентизиновой кислоты приводит к алкаптонурии. Гомогентизиновая кислота накапливается в крови, тканях и моче. Окисление и полимеризация этого вещества приводит к образованию пигмент» алкаптона, подобно тому как полимеризация ДОФА (см. рис. 42) приводит к образованию меланина. Отложение алкаптона в хрящах, которые затем темнеют, называют охронозом. В последующие годы он может вызвать артрит, а также заметное при клиническом обследовании потемнение ушей. Превращению гомогентизиновой кислоты в алкаптон способствует щелочная среда; при алкаптонурии наиболее явным симптомом является экскреция либо черной мочи, либо мочи, которая темнеет по мере защелачивания при хранении. Но во многих случаях этот симптом может отсутствовать. Часто первые признаки аномалии замечает мать, обеспокоенная темным цветом детских пеленок, которые становятся еще темнее при стирке с применением щелочного мыла или стиральных порошков. Указанная аномалия совместима с нормальной продолжительностью жизни и не требует лечения, но в среднем возрасте и позже обычно развивается артрит. Гомогентизиновая кислота — восстанавливающее вещество, реагирующее с таблетками клинитест. Алкаптонурия наследуется по аутосомнорецесспвному типу. Гетерозиготных носителей не удается обнаружить ни клиническими, ни биохимическими методами. Альбинизм. Недостаточность тирознпазы в меланощггах вызывает одну из форм альбинизма и наследуется как рецессивный признак. У пациентов отсутствует пигментация кожи, волос и радужной оболочки (глаза кажутся розовыми). Отсутствие пигментации кожи и радужной оболочки сопровождается острой фотосенсибилизацией. Тирозиназа, участвующая в биосинтезе катехоламинов, представляет собой другой фермент, контролируемый иным геном. У альбиносов метаболизм адреналина в норме. Нарушения обмена других аминокислот Болезнь «кленового сиропа». При болезни «кленового сиропа» происходит недостаточное декарбоксилирование оксокислот, образующихся в результате дезаминирования трех аминокислот с разветвленной цепью атомов углерода в молекуле: лейцина, изолейцина и валина. Эти аминокислоты накапливаются в крови и экскретируются (вместе с соответствующими оксокислотами) с мочой. В названии болезни отражен тот факт, что запах мочи больного напоминает запах кленового сиропа. Признаки заболевания проявляются в течение первой недели жизни. Если лечение не проводится, то в течение нескольких недель или месяцев развиваются тяжелые поражения нервной системы и наступает смерть. Но если заболевание диагностировано и назначена диета, бедная аминокислотами с разветвленной цепью атомов углерода, то возможно нормальное развитие ребенка. Диагноз ставится на основании повышенных уровней аминокислот с разветвлено!! цепью атомов углерода в крови и моче. Дефицит соответствующих ферментов в лейкоцитах может подтвердить диагноз. Заболевание наследуется по рецессивному типу. Гистидинемия. Гистидинемия связана с недостаточностью фермента гистидазы, участвующего в норме в метаболизме гистидина. При данном патологическом состоянии в крови повышена концентрация гистидина, а в моче увеличено содержание гистидина и его побочного метаболита имидазолпировиноградной кислоты. Подобно фенилиировиноградиои кислоте (экскретируемой при фенилкетонурии), имидазолпировиноградная кислота реагирует с хлорным железом, образуя синезеленый пигмент в присутствии хлорного железа или препарата фенистикса. Приблизительно в половине описанных в литературе случаев отмечают умственную отсталость, дефекты рсчп, у остальных больных заметных отклонений от нормы нет. Результаты диетотерапии, по современным данным, не представляются убедительными. По-видимому, заболевание наследуется по аутосомнорецессивному типу. Нарушения обмена металлов Два врожденных патологических состояния связаны с аномальным накоплением металлов в организме. Чрезмерная нагрузка железом (идиопатический гемохроматоз) обсуждается в гл. XVIII. Накопление меди происходит при болезни Уилсона. Болезнь Уилсона (гепатолентикулярная дегенерация) В норме большая часть меди плазмы крови входит в состав белка церулоплазмина, но некоторое ее количество образует непрочную связь с альбумином плазмы крови. Экскреция меди происходит преимущественно с желчью. При болезни Уилсона отмечают два нарушения метаболизма меди: 1) нарушение экскреции меди с желчью, что приводит к отложению меди в печени; 2) недостаточность церулоплазмина, результатом чего является низкое содержание меди в плазме крови. При этом медь преимущественно находится в непрочно связанном виде; ее отложение в тканях и фильтрация в почечных клубочках происходят более легко, чем в норме; экскреция меди с мочой повышена. Последствиями чрезмерного отложения меди в базальных ганглиях головного мозга, в печени, почечных канальцах, тканях глаза являются: 1) неврологические симптомы, обусловленные дегенерацией базальных ганглиев; 2) поражения печени, приводящие к циррозу; 3) поражения почечных канальцев с любыми (или всеми) биохимическими проявлениями этого патологического состояния, включая аминоацидурию (синдром Фанкони); 4) кольца Кайзера — Флейшера, окружающие роговую оболочку вследствие отложения меди в десцеметовые оболочки. Диагностика. У подавляющего большинства больных концентрации церулоплазмина и меди в плазме крови понижены. Интерпретируя результаты анализов содержания церулоплазмина, важно помнить, что низкая его концентрация возможна. также при следующих обстоятельствах: 1) в течение первых нескольких месяцев жизни; 2) при недостаточности питания; 3) при нефротическом синдроме в связи с потерями церулоплазмина с мочой. Повышенные концентрации церулоплазмина в крови могут быть в следующих случаях: 1) при острых патологических состояниях печени (что может объяснить редко наблюдаемый нормальный уровень церулоплазмина при болезни Уилсона); 2) в течение последнего триместра беременности; 3) у женщин, принимающих контрацептивные препараты; 4) при неспецифических поражениях печени (например, при воспалительных процессах или новообразованиях). В некоторых случаях диагностическое значение могут иметь результаты определений ссодержания меди в образцах печеночной ткани, полученных методом биопсии. Заболевание наследуется по рецессивному типу, но пониженные концентрации церулоплазмина могут быть у гетерозиготных носителей. Важно отличать их от гомозигот, у которых клинических симптомов заболеваний пока нет, хотя лечение следует проводить. Цель лечения с помощью таких образующих внутрпкомплексные соединения с медью веществ, как Dпеницилламин, — понижение тканевой концентрации меди. Лекарственные средства и врожденные аномалии обмена веществ Вариации индивидуальной чувствительности к идентичным лекарственным средствам отчасти могут быть предопределены генетически. Существует целый ряд хорошо изученных врожденных нарушений, которые под воздействием определенных лекарственных средств усугубляются или даже выявляются клинически. Подобные состояния можно разделить на две группы. Расстройства, обусловленные нарушениями метаболизма лекарств. Миорелаксант дитилин в норме оказывает весьма кратковременное действие, так как он быстро распадается в результате реакции, катализируемой холинэстеразой плазмы крови. При повышенной чувствительности к дитилину в организме имеется аномальная холинэстераза, распад лекарственного средства нарушен и может наступить длительный респираторный паралпч. Два других врожденных расстройства характеризуются нарушением метаболизма лекарственных средств изониазпда и дифенина соответственно. При этих расстройствах у больных отмечают токсические явления чаще и под воздействием более низких доз этих препаратов, чем у здоровых. Расстройства, обусловленные аномальным ответом на действие лекарств. Одна из форм гемолитической анемии, часто встречающаяся во многих регионах мира, обусловлена недостаточностью глюкозо6фосфатдегидрогеназы (Г6ФД) в эритроцитах. Указанный фермент — первый компонент гексозомонофосфатного шунта, необходимый для образования NADPH. В свою очередь NADPH, по-видимому, необходим для поддержания целостности биомембран эритроцитов. Описано множество вариантов недостаточности Г6ФД. Во многих случаях гемолиз усугубляли лекарственные средства, в частности, некоторые противомалярийные препараты, такие как примахин. а также сульфонамиды и аналоги витамина К. При врожденной печеночной порфирии острые приступы могут быть спровоцированы некоторыми лекарственными средствами, в частности барбитуратами. У некоторых лиц препараты, применяемые для общей анестезии (особенно часто фторотан в сочетании с дитилином), вызывают резкое повышение температуры тела, ригидность мышц и ацидоз; при этом большинство пациентов погибает (злокачественная гиперпирексия). Многие (но не все) члены семей этих больных имеют повышенный уровень креатинкиназы (КК). Этот краткий раздел должен напомнить читателям о возможности врожденной аномалии в тех случаях, когда имеется несвойственная норме реакция организма на воздействие лекарственного средства. В конце данной главы мы приводим ссылку на работу, характеризующую эту развивающуюся область фармакогенетики. ЗАКЛЮЧЕНИЕ 1. Врожденные нарушения обмена веществ могут вызывать заболевания, обусловленные наследуемыми дефектами биосинтеза белка. В большинстве случаев, когда выражены клинические симптомы, патологический процесс имеет в своей основе нарушения биосинтеза белка. 2. Врожденные нарушения обмена веществ могут не сопровождаться развитием клинических симптомов. Возможно появление клинических симптомов только при определенных обстоятельствах (например, при наличии вариантов холинэстеразы). С другой стороны, врожденные аномалии метаболизма могут вызывать тяжелые заболевания; некоторые из них смертельны. 3. Обнаружение определенных врожденных аномалий обмена веществ представляет только академический интерес. Диагностика наследуемых аномалий метаболизма имеет важное значение, если заболевание является тяжелым, но поддающимся лечению, если удается избежать воздействия факторов, усугубляющих течение заболевания, или если существуют трудности при дифференцировании с другими патологическими состояниями. 4. Наследование заболеваний может быть аутосомным или сцепленным с полом, доминантным или рецессивным. По характеру наследования патологические состояния, приводящие к развитию тяжелых клинических симптомов, преимущественно бывают аутосомнорецессивными; наиболее часто они встречаются в случаях брака между кровными родственниками. 5. Во многих случаях наследования клинически диагностируемого заболевания по рецессивному типу менее выраженные аномалии можно обнаружить с помощью биохимических тестов. 6. В этой главе рассмотрены некоторые врожденные нарушения метаболизма, не упомянутые в других разделах данной книги. СПИСОК ЛИТЕРАТУРЫ Holton J. В. Diagnosis of inherited metabolic diseases in severely ill children.— Ann. Clin. Biochem., 1982, 19, 389—395. Holtzman N. A. Newborn screening for inborn errors of metabolism. — Pediat, Clin. N. Amer., 1978, 25. 411— 421 Seriver С. R., Clow С. L. rheuvlkeloiniria: epitome of human biochemical genetics. — Now Engl. J. Med., I960, 303, 1336—1342 and 1394—140&. Vesell E. S. Pharmacogenetics: multiple interactions between genes and environment as determinants of drug response. — Amer. J. Med., 1979, 66, 183— 187. Stanbary J. В., Wyngaarden J. В., Fredrickson D. S., Goldstein J. L., Brown M. S.I Eds. The metabolic basis of inherited disease, 5th edit., New York: McGrawHill, 1983. НЕКОТОРЫЕ ВРОЖДЕННЫЕ НАРУШЕНИЯ ОБМЕНА И ИХ НАСЛЕДУЕМОСТЬ Приведенный ниже перечень врожденных нарушений метаболизма отнюдь не является полным. Он предназначен только для справок; не следует пытаться выучить этот список. Большинство из перечисленных аномалий мы обсуждаем в этой книге (соответствующая страница указана). За исключением тех случаев, когда подзаголовок относится к группе заболеваний с определенным типом наследуемое™, мы указываем характер наследования, если он известен. D—аутосомнодоминантный; Р — аутосомнорепессивный; Х-сцепленный; D—Х-сцепленный доминантный; Х-сцепленный; Р—Х-сцепленный рецессивный.
ГЛАВА XVII |