Клиническая химия в диагностике и лечении. Обмен натрия и воды обмен калия лечение диуретиками гомеостаз ионов водорода
 Скачать 3.99 Mb. Скачать 3.99 Mb.
|

ПОРФИРИИПорфирии представляют собой группу обычно врожденных расстройств синтеза гема. Хотя они, как правило, не имеют широкого распространения, некоторые считаются очень редкими заболеваниями, их распознавание и обследование родственников больного представляются важными задачами. Так, при наследственных печеночных порфириях необходимо избегать контактов с лекарственными средствами, которые могут провоцировать острые приступы, закапчивающиеся иногда трагическим исходом. ФИЗИОЛОГИЯ Гем синтезируется в большинстве тканей нашего организма. В костном мозге гем включается в гемоглобин. В других клетках тем используется при синтезе цитохромов и родственных им соединений. Цитохромы — ото компоненты цепи переноса электронов, которая необходима для адекватного использования энергии, генерируемой в процессах обмена веществ. Поэтому нарушение синтеза цитохромов может иметь важные последствия. Количественно печень — самый большой орган, в котором образование гема не связано с эритропоэзом. Биосинтез гема. Ниже и на рис. 46 перечислены основные этапы синтеза гема. 5-Аминолевулинат (АЛ) образуется путем конденсации глицина и сукцината. Для осуществления этой реакции, катализируемой ферментом АЛсинтазой, требуется пиридоксальфосфат. Две молекулы АЛ конденсируются, образуя монопиррол порфобилиноген (ПБГ). Взаимодействие 4 молекул ПБГ приводит к образованию тетрапиррола уропорфириногепа (рис. 47). При этом образуются два изомера, I и III. Изомер III вовлечен в главный путь обмена, приводящий к образованию гема путем последовательного синтеза копропорфириногена и протопорфирина с последующим включением железа. Каждый из указанных этапов контролируется определенным ферментом. В норме скорость процесса синтеза гема в целом ограничивает реакция, катализируемая АЛсинтазой; регуляция этого синтеза осуществляется его конечным продуктом гемом по принципу обратной связи. Порфириногены и их предшественники (АЛ и ПБТ) — бесцветные соединения. Однако порфириногены подвергаются спонтанному окислению до соответствующих порфиринов, которые имеют темнокрасную окраску и флуоресцируют в ультрафиолетовом свете. При аэробных условиях и на свету возможно также спонтанное образование уропорфирина из ПБГ. При хранении мочи, содержащей большие количества порфириногенов или их предшественников, она постепенно темнеет. 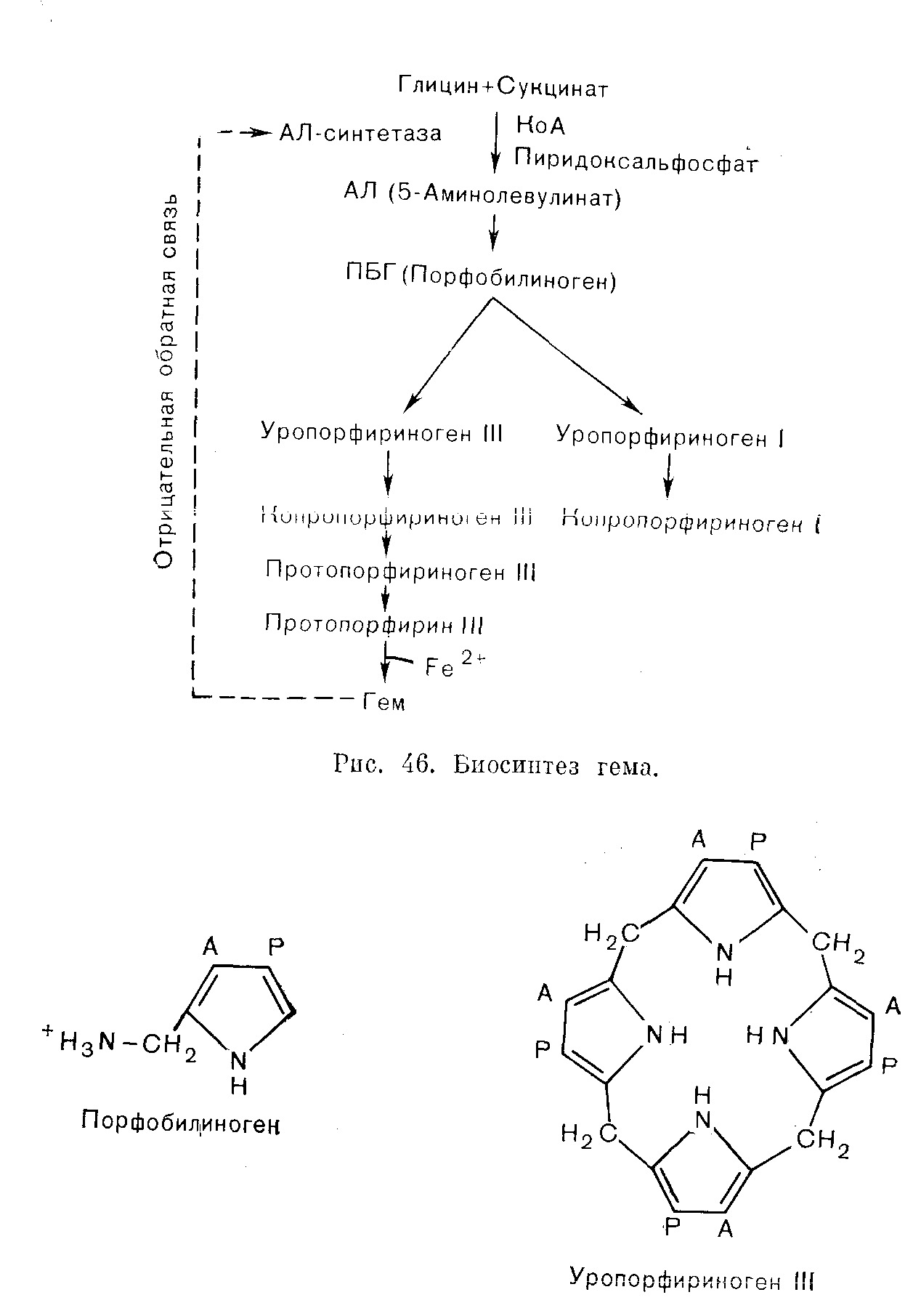 Рис. 47. Порфобилиноген и тетрапиррол уропорфирипогеп III, в молекуле которого имеется 4 остатка порфобилиногена. Уроиорфириноген I отличается только порядком расположения боковых цепей, присоединенных к одной из циклических структур. (Боковые цепи: А — ацетат; Р — пропионат). Экскреция. Любой избыток промежуточных продуктов синтеза гема экскретируется. В моче появляются такие растворимые в воде соединения, как АЛ, ПБГ и уропорфирин (оген). Протопорфирин экскретируется в желчь и появляется в фекалиях. Копропорфирин (оген) может быть экскретирован любым путем. По мере нарастания щелочности мочи повышается выведение через почки копропорфириногена, а также уробилиногена. Тесты, применяемые для скрининга (см. ниже), не позволяют обнаружить имеющиеся в моче здорового человека концентрации АЛ, ПБГ ii порфирина. Фекалии могут содержать порфирин в концентрациях, достаточных для появления слабой флуоресценции в экстрактах. ПОРФИРИИ Порфирии обусловлены недостаточностью одного ыз ферментов, участвующих в образовании гема; следовательно, синтез гема нарушается. Сниженное ингибирование АЛсинтазы (по принципу обратной связи) может поддерживать адекватные уровни гема, но только за счет образования избытка порфиринов или их предшественников. Симптомы порфирии хорошо коррелируют с биохимическими аномалиями. Поражения кожи, варьирующие от легкой фотосенсибилизации до обширного образования волдырей, наблюдают при чрезмерном синтезе порфиринов. Типична локализация поражений на открытой поверхности кожи, где солнечные лучи вызывают активацию порфиринов с высвобождением энергии, повреждающей ткани. Нейрологические расстройства, такие как периферические невриты, боли в области живота и их сочетания (при острых приступах), бывают лишь при порфириях, протекающих на фоне чрезмерного синтеза предшественников — АЛ и ПБГ. Не установлено, обусловлены ли эти нейрологические расстройства недостаточностью гема в нервной системе или непосредственным токсическим действием АЛ или ПБГ. Обычно порфирии классифицируют в зависимости от того, накапливаются ли порфирины преимущественно в печени или в эритропоэтической системе. Эти особенности локализации порфиринов не следует рассматривать как безусловное указание на наличие недостаточности соответствующего фермента только в данной системе. Печеночные порфирии. Различают острые порфирии (острая перемежающаяся порфирия, porphyria variegata, и врожденная копропорфирия) и porphyria cutanea tarda (генетически детерминированная и приобретенная). Эритропоэтические порфирии. Различают врожденную эритропоэтическую порфирию и эритропеченочную протопорфирию. Особенности порфирии указаны в табл. 32 и кратко рассмотрены ниже. Острые печеночные порфирии Острая перемежающаяся порфирия, врожденная копропорфирия и porphyria variegata представляют собой патологические состояния, наследуемые по доминантному типу. В каждой из этих трех болезнен различают латентную и острую фазы. Симптомы и биохимические аномалии, характерные для латентных фаз, различны; они соответствуют природе фермента, недостаточность которого имеет место. Однако во время острых фаз развиваются Таблица 32. Основные клинические и биохимические особенности порфирии 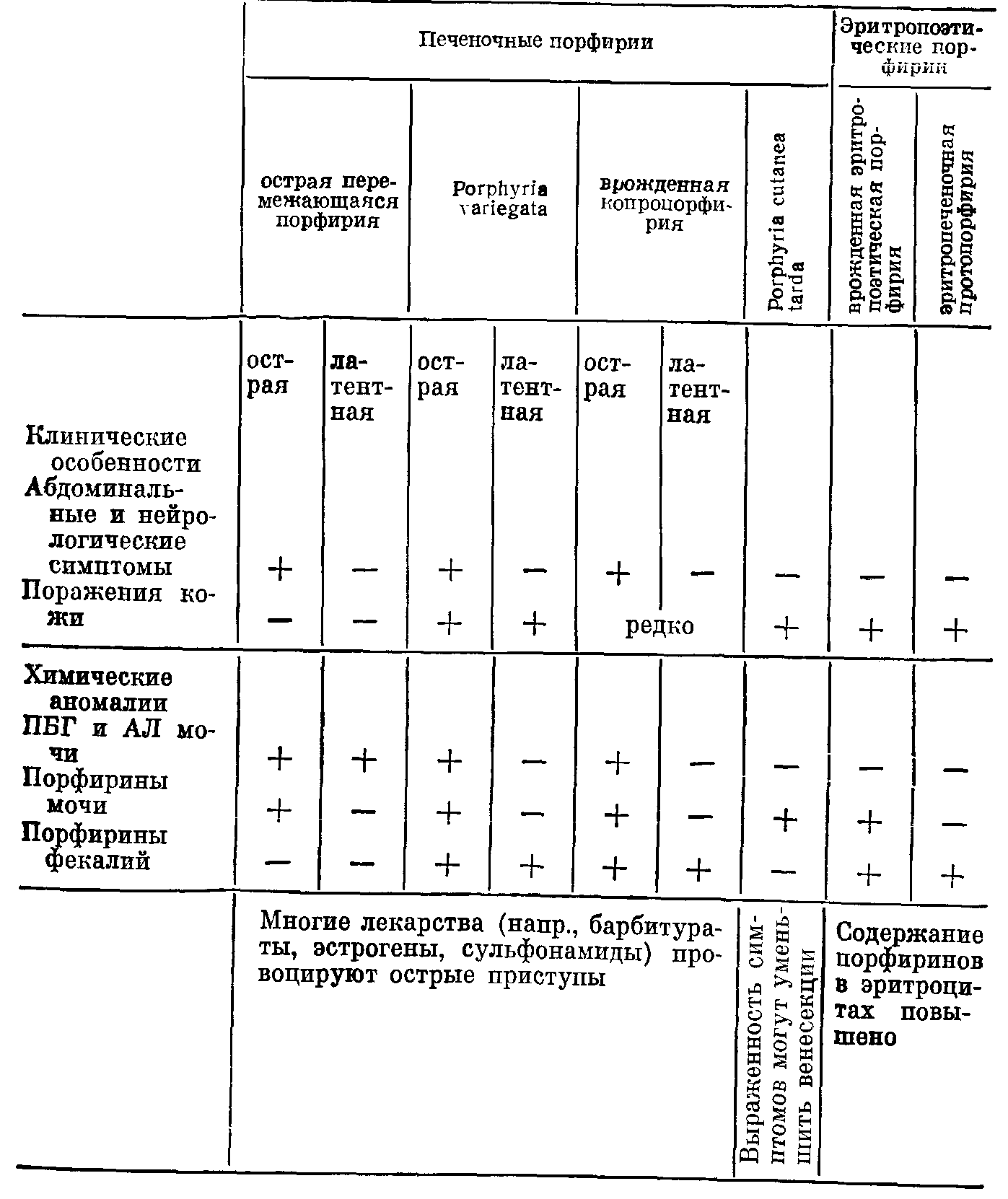 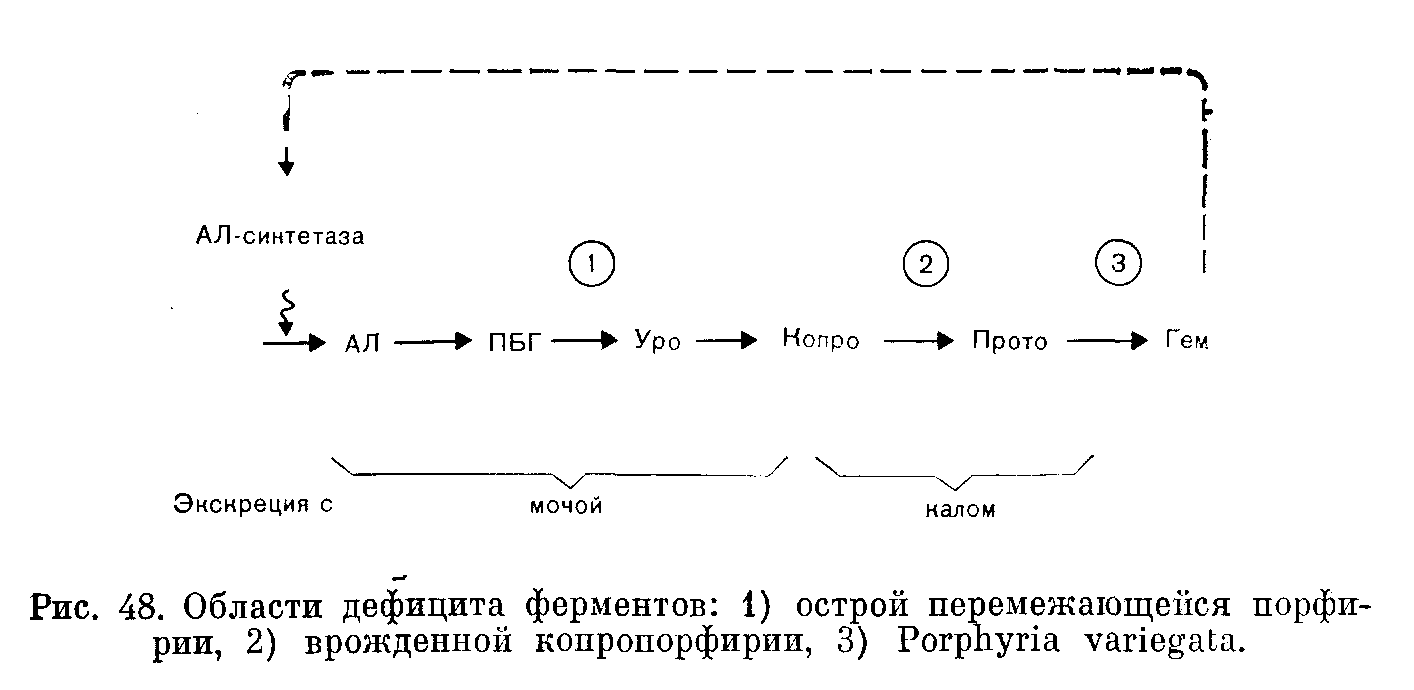 связанные с нейрологическими и абдоминальными симптомами биохимические и клинические аномалии, характерные для чрезмерной экскреции АЛ и ПБГ. Черты сходства и различия между отдельными порфириями легче объяснить, если обратиться к упрощенной схеме путей синтеза гема (рис. 48). Латентная фаза. Недостаточность ферментов приводит к уменьшению содержания гема, что в свою очередь способствует повышению активности АЛсинтазы (по принципу отрицательной обратной связи). Показано, что при всех порфириях активность АЛсинтазы повышена. Благодаря этому поддерживается постоянная концентрация гема (за счет накопления и экскреции соединений, расположенных непосредственно перед пунктами блокирования). Основные биохимические аномалии предсказуемы (рис. 48): острая перемежающаяся порфирия — увеличение содержания АЛ и ПБГ в моче (обнаруживается не у всех больных пациентов); врожденная копропорфирия—увеличение содержания копропорфирина в фекалиях; porphyria variegata — увеличение содержания протопорфирина в фекалиях. При острой перемежающейся порфирии латентная фаза обычно бессимптомна. При porphyria variegata, а также (реже) при врожденной копропорфирии повышение концентрации порфиринов может вызывать поражения кожи. Острая фаза. Дальнейшее повышение активности АЛсинтазы может быть вызвано воздействиями целого ряда лекарственных средств (в частности барбитуратов, эстрогетюв, сульфопамидов и гризеофульвина) и острыми патологическими процессами и может быть либо прямым эффектом, либо следствием возрастания потребности в геме. В результате происходит значительное увеличение экскреции АЛ и ПБГ. При острой перемежающейся порфирии это увеличение обусловлено блокированием, возникающим в результате врожденной недостаточности уропорфириноген1синтазы; при печеночной копропорфирии и porphyria variegata этот фермент становится ограничивающим скорость процесса в целом и теряющим свойство адекватно реагировать на возрастание потребности в геме. Во время приступов острой порфирии особенно важное значение имеет увеличение экскреции с мочой АЛ и ПБГ, проявляющееся (на фоне других биохимических аномалий) и при врожденной копропорфирии или porphyria variegata. Повышение скорости реакций этого пути обмена веществ и спонтанное превращение предшественников в порфирины приводит к увеличению их экскреции с мочой. Клиническими симптомами при острых приступах порфирии обычно являются имеющие характер колик боли в области живота (вследствие вовлечения автономной нервной системы) и нейрологические расстройства, варьирующие от периферических невритов до квадриплегии. Смерть может наступить от респираторного паралича. Часто отмечают гипонатриемию. Острые приступы бывают только после достижения половой зрелости и чаще у женщин, чем у мужчин. Острый приступ порфирии весьма напоминает симптомы, характерные для тяжелых острых абдоминальных патологических процессов. Если диагноз не поставлен, больного могут прооперировать с применением барбитуратов в качестве анестезирующих средств; барбитураты и стресс, связанный с операцией, ухудшат состояние больного. Диагностика латентной порфприи. Необходимо обследовать родственников всех больных, страдающих порфпрпей. Применяемые для скрининга тесты на избыток ПБГ и АЛ в моче неадекватны для диагностики латентной острой перемежающейся порфирии; даже количественное определение ПБГ и АЛ может не обнаружить всех носителей этого заболевания. Наиболее доступные клетки, в которых следует определять активность фермента уропорфириноген1сиптазы, — эритроциты. Уровни активности у больных и здоровых часто перекрываются, но в пределах семьи носители заболевания обнаруживают активность, приблизительно в 2 раза меньшую, чем их здоровые родственники. С помощью этого теста можно также выявить пораженных заболеванием детей. Носителей porphyria variegata и врожденной копропорфирии можно идентифицировать после достижения ими полового созревания путем обнаружения отчетливо повышенной экскреции порфнрпнов с фекалиями. Положительные результаты скрининговых тестов следует подтвердить количественными определениями. Porphyria cutanea tarda (кожнопеченочная порфирия) В противоположность острой перемежающейся порфирии porphyria cutanea tarda не сопровождается обострениями патологического процесса, и выраженность ее симптомов не изменяется после приема внутрь барбитуратов. Слабо выраженные проявления копропорфиринурии могут быть у больных с заболеваниями печени; реже чрезмерный синтез и экскреция уропорфирина (главным образом типа I) сопровождаются резкой фотосенсибилизацией и поражениями кожи (porphyria cutanea tarda). В случаях с хроническим течением заболевания наблюдают гирсутизм и гиперпигментацию. Вероятно, у большинства больных имеется наследуемая по доминантному типу недостаточность фермента уропорфириногендекарбоксилазы, превращающего уропорфириноген в копропорфириноген. Клинические симптомы заболевания, однако, проявляются только после того как дополнительный фактор усугубляет имеющуюся недостаточность фермента. Роль такого фактора могут играть заболевания печени, обусловленные злоупотреблением алкоголя или перегрузкой железом, а также лечение высокими дозами эстрогеыов. Еслн зги факторы исключить, сенсибилизация кожи уменьшается. Сходные симптомы могут вызывать такие действующие на печень токсические вещества, как гексахлорбензол, который может непосредственно угнетать активность указанного фермента. Эритропоэтические порфирии С накоплением порфиринов в эритроцитах связаны 2 редких наследственных заболевания, для которых приступы острой порфирии не характерны, а экскреция АЛ и ПБГ в пределах нормы. Врожденная эритропоэтическая порфирия в отличие от всех других порфирии, рассмотренных выше, наследуется как рецессивный признак. Обычно с самого детства и далее на протяжении жизни больного концентрации уропорфирина I в эритроцитах и плазме крови очень высоки, что сопровождается резкой фотосенсибилизацией. Порфирины накапливаются также в костях и зубах, которые флуоресцируют в ультрафиолетовом свете п могут иметь коричневаторозовую окраску. Кроме того, отмечают гирсутизм (особенно на лице) и гемолитическую анемию. Резко повышена экскреция порфиринов с мочой, а также (в меньшей степени) с фекалиями. Эритропеченочная протопорфирия. При этом наследуемом по доминантному типу заболевании содержание протопорфирина повышено в эритроцитах и в фекалиях. Имеется умеренная фотосенсибилизация; поражение клеток печени может приводить к ее недостаточности. Другие причины экскреции избытка порфиринов Порфирия не является единственной причиной нарушенного метаболизма порфиринов. Положительные результаты используемых при скрининге тестов должны быть подтверждены количественным анализом с идентификацией экскретируемых порфиринов. Следует помнить о трех причинах. Отравление свинцом ингибирует несколько ферментов, участвующих в синтезе гема, и вызывает в конечном счете анемию. Моча содержит повышенные концентрации АЛ (ранний и чувствительный тест) и копропорфирина. Некоторые симптомы отравления свинцом (такие как боль в области живота) сходны с симптомами острого приступа порфирии, что может вызывать затруднения при дифференциальной диагностике. Заболевания печени могут приводить к увеличению содержания копропорфирина в моче, по-видимому, в связи с его пониженной экскрецией с желчью. По-видимому, это самая частая причина порфиринурии. В некоторых случаях развивается умеренная фотосенсибилизация (при porphyria cutanea tarda более тяжелые поражения кожи обусловлены воздействием избытка уропорфирина). Язвенные поражения грпкспмллт.ных отделов желучочяокишечного тракта могут приводить к увеличению содержания порфиринов в фекалиях в связи с распадом гемоглобина. Если в дистальных отделах пищеварительного тракта есть кровотечение, то кровь достигает прямой кишки прежде, чем успевают произойти соответствующие превращения; эти сведения могут помочь в приблизительной локализации кровотечения. ЗАКЛЮЧЕНИЕ 1. Порфирины—побочные продукты синтеза гема; АЛ и ПБГ — предшественники. 2. Порфириями называют заболевания, связанные обычно с врожденными нарушениями метаболизма порфиринов (табл. 32). 3. Для врожденных печеночных порфирии характерны острыеприступы с абдоминальными и нейрологическими симптомами. Потенциально такие приступы опасны для жизни. Многие лекарственные средства могут провоцировать эти приступы. Диагностика порфирии в острой фазе основана на обнаружении АЛ и ПБГ в моче. 4. Если поставлен диагноз врожденных порфирии, обследуют всех родственников больного для выявления бессимптомных случаев (моча, фекалии). Скрининговые тесты могут дать отрицательные результаты при некоторых типах порфирии, в связи с чем необходимы количественные определения. 5. Другие причины аномалий в экскреции порфиринов — отравление свинцом, заболевания печени, кровотечения в проксимальных отделах желудочнокишечного тракта. 6. Очень редкие Эритропоэтические порфирии приводят к накоплению избытка порфиринов в эритроцитах. ТЕСТЫ ДЛЯ СКРИНИНГА НА ПОРФИРИИ Обычно тесты для скрининга на порфирии выполняют в лаборатории. Необходимым условием является наличие: 1) источника ближнего ультрафиолетового света (лампа Вуда); 2) растворителя для экстракции (смесь равных частей этилового эфира, ледяной уксусной кислоты и амилового спирта); 3) реагента Эрлиха (2% пдиметиламинобензальдегид в 5 М НС1); 4) пбутанола. Тест на порфобилипотен. 1. Смешивают равные части свежей мочи и реагента Эрлиха. Если в пробе содержится ПБТ, развивается красное окрашпванне. 2. Еслц появилось красное окрашивание, добавляют 3 мл пбутанола, пробирку встряхивают и оставляют стоять до разделения фаз. Если красное окрашивание не переходит в бутанол (верхний слой), то эта окраска обусловлена наличием ПБГ. Важно использовать свежую мочу, поскольку при ео хранении ПБГ исчезает. Целый ряд других соединений, особенно уробилиногеп, могут также давать краской окрашивание с реагсшом Орлиха. Все известные соединения этого типа, однако, извлекаются в бутаноловый слой. Если вместо бутанола взять хлороформ, то при этом могут получиться ложноположительные результаты, поскольку не все отличающиеся от ПБГ соединения экстрагируются в органическую фазу. Тест на порфирнны. М о ч а. Приблизительно 10 мл свежей мочи смешивают с 2 мл растворителя (п. 2), экстрагирующего порфирины, и оставляют для разделения фаз. Красная флуоресценция верхнего слоя в ультрафиолетовом свете указывает на наличие порфиринов. фекалии. Кусочек кала размером с горошину смешивают с 2 мл растворителя. Красная флуоресценция в ультрафиолетовом свете указывает на наличие либо порфпринов, либо хлорофилла (имеющего сходную структуру и поступающего с пищей). Их можно различить, добавляя 1,5 М НС1, которая экстрагирует порфирины, но не хлорофилл. Интерпретация. Важны следующие моменты: 1. Существенное значение имеет адекватность тестируемого материала (моча пли фекалии) с учетом типа предполагаемой порфирии. 2. Чувствительность тестов, применяемых для скрининга, слишком низка для диагностики острой перемежающейся порфирии в латентной фазе. При правильном выполнении отрицательный результат тестов на порфирины фекалий почти однозначно исключает диагноз porphyria variegata. 3. До наступления половой зрелости тесты дают отрицательные результаты даже у детей, которые будут страдать порфирией. 4. Положительные результаты тестирования должны подтверждаться количественным анализом и путем идентификации порфиринов. Помимо порфирии, существуют другие причины повышения экскреции порфиринов. ГЛАВА XX |