ферменты1_razdel_1-23. Понятие фермент. Свойства ферментов. Отличие ферментов от неорганических катализаторов. Ферменты
 Скачать 1.94 Mb. Скачать 1.94 Mb.
|

ИЗОФЕРМЕНТЫИзоферменты, или изоэнзимы,– это множественные формы фермента, катализирующие одну и ту же реакцию, но отличающиеся друг от друга по физическим и химическим свойствам, в частности по сродству к субстрату, максимальной скорости катализируемой реакции (активности), электрофоретической подвижности или регуляторным свойствам. 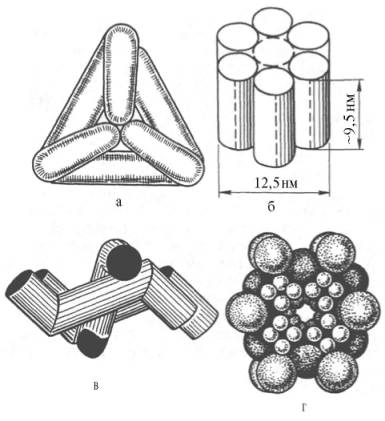 Рис. 4.5. Модели строения некоторых олигомерных ферментов. а - молекула глутаматдегидроге-назы, состоящая из 6 протоме-ров (общая мол. м. 336000); б -молекула РНК-полимеразы; в -половина молекулы каталазы; г - молекулярный комплекс пируватдегидрогеназы. В живой природе имеются ферменты, молекулы которых состоят из двух и более субъединиц, обладающих одинаковой или разной первичной, вторичной или третичной структурой. Субъединицы нередко называют протомерами, а объединенную олигомерную молекулу – мультимером (рис. 4.5; см. главу 1). Считают, что процесс олигомеризации придает субъединицам белков повышенную стабильность и устойчивость по отношению к действию денатурирующих агентов, включая нагревание, влияние протеиназ и др. Однако на нынешнем этапе знаний нельзя ответить однозначно на вопрос о существенности четвертичной структуры для каталитической активности ферментов, поскольку пока отсутствуют методы, позволяющие в «мягких» условиях разрушить только лишь четвертичную структуру. Применяемые обычно методы жесткой обработки (экстремальные значения рН, высокие концентрации гуанидинхлорида или мочевины) приводят к разрушению не только четвертичной структуры, но и вторичной и третичной структур стабильного олигомерного фермента, протомеры которого оказываются денатурированными и, как следствие этого, лишенными биологической активности. Следует указать на отсутствие ковалентных, главновалентных связей между субъединицами. Связи в основном являются нековалентными, поэтому такие ферменты довольно легко диссоциируют на протомеры. Удивительной особенностью таких ферментов является зависимость активности всего комплекса от способа упаковки между собой отдельных субъединиц. Если генетически различимые субъединицы могут существовать более чем в одной форме, то соответственно и фермент, образованный из двух или нескольких типов субъединиц, сочетающихся в разных количественных пропорциях, может существовать в нескольких сходных, но не одинаковых формах. Подобные разновидности фермента получили название изоферментов (изоэнзимов или, реже, изозимов). В частности, если фермент состоит из 4 субъединиц двух разных типов – Н и М (сердечный и мышечный), то активный фермент может представлять собой одну из следующих комбинаций: НННН, НННМ, ННММ, НМММ, ММММ, или Н4, Н3М, Н2М2, НМ3, М4, соответствующую изоферментам ЛДГ1, ЛДГ2, ЛДГ3, ЛДГ4 и ЛДГ5. При этом синтез Н- и М-типов осуществляется различными генами и в разных органах экспрессируется по-разному. В одних случаях субъединицы имеют почти идентичную структуру и каждая содержит каталитически активный участок (например, β-галакто-зидаза, состоящая из 4 субъединиц). В других случаях субъединицы оказываются неидентичными. Примером последних может служить трипто-фансинтаза, состоящая из 2 субъединиц, каждая из которых наделена собственной (но не основной) энзиматической активностью, однако, только будучи объединенными в макромолекулярную структуру, обе субъединицы проявляют триптофансинтазную активность. Термин «множественные формы фермента» применим к белкам, катализирующим одну и ту же реакцию и встречающимся в природе в организмах одного вида. Термин «изофермент» применим только к тем множественным формам ферментов, которые появляются вследствие генетически обусловленных различий в первичной структуре белка (но не к формам, образовавшимся в результате модификации одной первичной последовательности). Одним из наиболее изученных 4 ферментов, множественность форм которого детально изучена методом гель-электрофореза, является ЛДГ, катализирующая обратимое превращение пировиноградной кислоты в молочную. Пять изоферментов ЛДГ образуются из 4 субъединиц примерно одинакового размера, но двух разных типов. Поскольку Н-протомеры несут более выраженный отрицательный заряд при рН 7,0–9,0, чем М-про-томеры, изофермент, состоящий из 4 субъединиц Н-типа (Н4), при электрофорезе будет мигрировать с наибольшей скоростью в электрическом поле к положительному электроду (аноду). С наименьшей скоростью будет продвигаться к анодуизофермент М4, в то время как остальные изо-ферменты будут занимать промежуточные позиции. Следует подчеркнуть, что изоферменты ЛДГ, обладая почти одинаковой ферментативной активностью, различаются некоторыми физико-химическими свойствами: молекулярной массой, электрофоретической подвижностью, отношением к активаторам и ингибиторам и др., однако для каждой ткани в норме характерно свое соотношение форм (изоферментный спектр) ЛДГ. Например, в сердечной мышце преобладает Н4, т.е. ЛДГ1 , а в скелетных мышцах и печени – М4 (ЛДГ5) (рис. 4.6). Эти обстоятельства широко используют в клинической практике, поскольку изучение появления изоферментов ЛДГ (и ряда других ферментов) в сыворотке крови может представлять интерес для дифференциальной диагностики органических и функциональных поражений органов и тканей. По изменению содержания изоферментов в сыворотке крови можно судить как о топографии патологического процесса, так и о степени поражения органа или ткани. 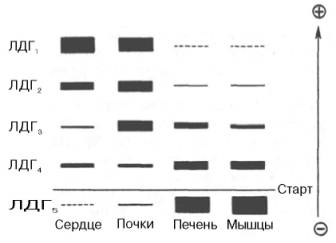 Рис. 4.6. Распределение и относительные количества изоферментов ЛДГ в различных органах. Экстракты нанесены на линию, отмеченную надписью «Старт». При заданных условиях опыта (рН) 4 изофермента ЛДГ движутся к аноду, а один (ЛДГ5) – к катоду. Красным цветом выделены основные изоформы ЛДГ для данного органа. 9. Современные представления о механизме действия ферментов и регуляции их активности, привести примеры. Структура и функции ферментов, а также механизм их действия почти ежегодно подробно обсуждаются на многих международных симпозиумах и конгрессах. Важное место отводится рассмотрению структуры всей молекулыфермента и ее активных центров, молекулярному механизму действия различных типов ферментов, общей теории энзиматического катализа. Тем не менее до сих пор нет полной ясности по двум кардинальным проблемам энзимологии: чем вызваны специфичность действия и высокая каталитическая эффективность ферментов? До установления химической природы ферментов гипотезы о механизме их действия опирались на исследования кинетики и модельные опыты химического гомогенного катализа. Повышение скорости химических реакций под действием ферментов объясняли следующим: а) активированием субстрата в результате образования адсорбционных или молекулярных, обратимо диссоциирующих фермент-субстратных комплексов; б) цепным механизмом реакций с участием радикалов или возбужденных молекул. Оказалось, что цепные механизмы реакциине играют существенной роли в биологическом катализе. После установления химической природы ферментовподтвердилось представление, выдвинутое более 80 лет назад В. Анри, Л. Михаэлисом и М. Ментен, о том, что при энзиматическом катализе фермент Е соединяется (в принципе обратимо) со своим субстратом S, образуя нестойкий промежуточный фермент-субстратный комплекс ES, который в конце реакции распадается с освобождением фермента и продуктов реакции Р. Благодаря высокому сродству связывания и образованию ES-комплекса резко возрастает число молекул субстрата, вступающих в реакции. Эти представления легли в основу теории «ключа-замка» Э. Фишера, которую иногда называют теорией «жесткой матрицы». Таким образом, жесткая структура активного центра оказывается комплементарной молекулярной структуре субстрата, обеспечивая тем самым высокую специфичность фермента. Л. Михаэлис не только постулировал образование промежуточного фермент-субстратного ES-комплекса, но и рассчитал влияние концентрации субстрата на скорость реакции. В процессе реакции различают несколько стадий: присоединение молекулы субстрата к ферменту, преобразование первичного промежуточного соединения в один или несколько последовательных (переходных) комплексов и протекающее в одну или несколько стадий отделение конечных продуктов реакции от фермента. Это можно схематически проиллюстрировать следующими примерами: В реакциях анаболизма, например А + В —> АВ, фермент может соединяться как с одним, так и с другим субстратомили обоими субстратами: 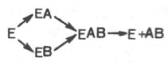 В реакциях катаболизма, например АВ —> А + В: 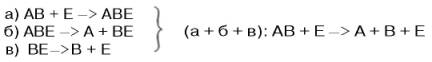 На рис. 4.7 представлена схема образования промежуточного фермент-субстратного комплекса. Если фермент в активном центре содержит кофермент, то предполагается образование тройного комплекса (рис. 4.8). Фермент вступает во взаимодействие с субстратом на очень короткий период, поэтому долгое время не удавалось показать образование такого комплекса. Прямые доказательства существования фермент-субстратного комплекса были получены в лабораториях Д. Кейлина и Б. Чанса. В настоящее время экспериментальные и математические методы кинетики, термодинамики и статической механики химических реакций позволяют определить для ряда ферментативных реакций кинетические и термодинамические показатели, в частности константы диссоциации промежуточных фермент-субстратных комплексов, константы скорости и равновесия их образования. 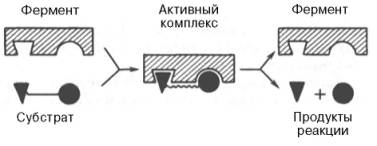 Рис. 4.7. Образование нестойкого фермент-субстратного комплекса согласно теории Э. Фишера «ключ-замок». 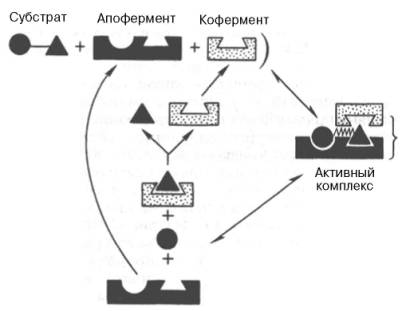 Рис. 4.8. Функция кофер-мента (по А. Кантарову и Б. Шепартцу). В образовании фермент-субстратных комплексов участвуют водородные связи, электростатические и гидрофобные взаимодействия, а в ряде случаев также ковалентные, координационные связи (рис. 4.9). Информация о природе связей между субстратом и связывающим участком активного центра фермента может быть получена методами ЭПРи ЯМР, а также методами УФ- и ИК-спектроскопии. Для каталитической активности фермента существенное значение имеет пространственная структура, в которой жесткие участки α-спиралей чередуются с гибкими, эластичными линейными отрезками, обеспечивающими динамические изменения белковой молекулы фермента. Этим изме-неням придается большое значение в некоторых теориях ферментативного катализа. Так, в противоположность модели Э. Фишера «ключ-замок» Д. Кошлендом была разработана теория «индуцированного соответствия», допускающая высокую конформационную лабильность молекулы белка-фермента и гибкость и подвижность активного центра. Эта теория была основана на весьма убедительных экспериментах, свидетельствующих о том, что субстрат индуцирует конформационные изменения молекулы фермента таким образом, что активный центр принимает необходимую для связывания субстратапространственную ориентацию. Иными словами, фермент только в присутствии (точнее, в момент присоединения) субстрата будет находиться в активной (напряженной) Т-форме в отличие от неактивной R-формы (рис. 4.10). На рис. 4.10 видно, что присоединение субстрата S к ферменту Е, вызывая соответствующие изменения конформацииактивного центра, в одних случаях приводит к образованию активного комплекса, в других – неактивного комплекса вследствие нарушения пространственного расположения функциональных групп активного центра в промежуточном комплексе. Получены экспериментальные доказательства нового положения о том, что постулированное Д. Кошлендом «индуцированное соответствие» субстрата и фермента создается не обязательно изменениями конформации белковой молекулы, но также геометрической и электронно-топографической перестройкой молекулы субстрата. 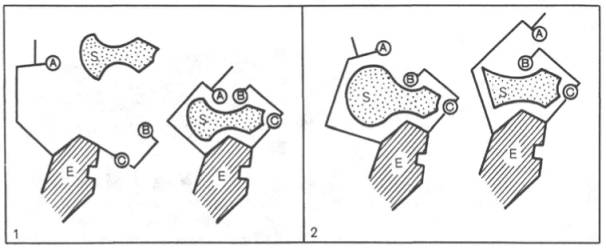 Рис. 4.10. Изменения структуры активного центра фермента, вызванные субстратом, согласно модели «индуцированного соответствия» Д. Кошленда. А, В, С - функциональные группы активного центра; 1 - активный комплекс; 2 - неактивный комплекс. 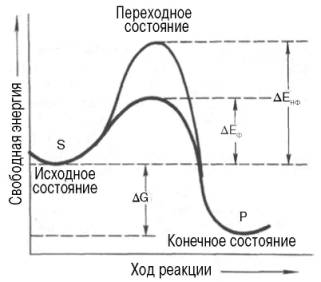 Рис. 4.11. Энергетический механизм ферментативной и неферментативной химических реакций. S - исходный субстрат; Р - продукт; ΔЕНФ -энергия активации неферментативной реакции; ΔЕФ - энергия активацииферментативной реакции; ΔG - стандартное изменение свободной энергии. В каталитическом процессе существенное значение имеют точное соответствие между ферментом и субстратом, а также термодинамические и каталитические преимущества подобного соответствия. Гипотеза «индуцированного соответствия» предполагает существование между ферментом и субстратом не только пространственной или геометрической компле-ментарности, но и электростатического соответствия, обусловленного спариванием противоположно заряженных групп субстрата и активного центра фермента. Точное соответствие обеспечивает образование эффективного комплекса между субстратом и ферментом. Подобно другим катализаторам, ферменты, с термодинамической точки зрения, ускоряют химические реакции за счет снижения энергии активации . Энергией активации называется энергия, необходимая для перевода всех молекулмоля вещества в активированное состояние при данной температуре. Другими словами, это энергия, необходимая для запуска химической реакции, без которой реакция не начинается несмотря на ее термодинамическую вероятность. Фермент снижает энергию активации путем увеличения числа активированных молекул, которые становятся реакционноспособными на более низком энергетическом уровне (рис. 4.11). На рисунке видно, что ферментативная реакция имеет более низкую энергию активации. Следует отметить, что как катализируемая ферментом, так и не катализируемая им реакция независимо от ее пути имеет одинаковую величину стандартного изменения свободной энергии (ΔG). Действуя на скорость реакции, ферменты не изменяют равновесия между прямой и обратной реакциями, как и не влияют на величину свободной энергии реакции; они лишь ускоряют наступление равновесия химической реакции. Зависимость между константой равновесия и изменением свободной энергии реагирующих веществ математически принято выражать уравнением ΔG = = –R•T•lnK, где R – газовая постоянная; Т – абсолютная температура в Кельвинах; lnК – натуральный логарифм константы равновесия; ΔG – стандартное изменение свободной энергии, Дж/моль. Из представленного уравнения вытекает, что при высоком значении К величина ΔG оказывается отрицательной. Подобные реакции сопровождаются уменьшением свободной энергии. При низком значении К величина ΔG оказывается положительной. Если константа равновесия равна единице, то изменение свободной энергии будет равно нулю и реакция легкообратима. Для измерения константы равновесия и величины свободной энергии какой-либо химической реакции, например реакции взаимопревращения глюкозо-1-фосфата в глюкозо-6-фосфат, катализируемой ферментомфосфоглюкомутазой, определяют количество глюкозо-6- и глюкозо-1-фосфата при достижении химического равновесия. В состоянии равновесия содержание глюкозо-6-фосфата оказывается в 19 раз больше количества глюкозо-1-фосфата. Отсюда константа равновесия К равна 19. Подставляя эту цифру в уравнение, получаем ΔG = –7329 Дж/моль. Это означает, что при превращении 1 моля глюкозо-1-фосфата в 1 моль глюкозо-6-фосфата при температуре 25°С происходит уменьшение свободной энергии системы на 7329 Дж. Таким образом, в механизме ферментативного катализа ведущую роль играют промежуточные фермент-субстратные комплексы, образование которых определяется как тонкой трехмерной структурой активного центра, так и уникальной структурной организацией всей молекулы фермента, обеспечивающими высокую каталитическую активность и специфичность действия биокатализатора. |