ферменты1_razdel_1-23. Понятие фермент. Свойства ферментов. Отличие ферментов от неорганических катализаторов. Ферменты
 Скачать 1.94 Mb. Скачать 1.94 Mb.
|

|
10. Современные представления о кинетике ферментативных реакций и факторы, которые на нее влияют. Одним из характерных проявлений жизни является удивительная способность живых организмов кинетически регулировать химические реакции, подавляя стремление к достижению термодинамического равновесия. Ферментативная кинетика занимается исследованием закономерностей влияния химической природы реагирующих веществ (ферментов, субстратов) и условий их взаимодействия (концентрация, рН среды, температуры, присутствие активаторов или ингибиторов) на скорость ферментативной реакции. Главной целью изучения кинетики ферментативных реакций является получение информации, которая может способствовать выяснению молекулярного механизма действия фермента. Общие принципы кинетики химических реакций применимы и к ферментативным реакциям. Известно, что любая химическая реакция характеризуется константой термодинамического равновесия. Она выражает состояние химического равновесия, достигаемого системой, и обозначается Кр. Так, для реакции: 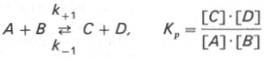 константа равновесия равна произведению концентраций образующихся веществ, деленному на произведение концентрации исходных веществ. Значение константы равновесия обычно находят из соотношения констант скоростей прямой (k+1) и обратной (k– 1 ) реакций, т.е. Кp = k+1/k–1. В состоянии равновесия скорость прямой реакции: v+1 = k + 1[ А ] • [ B ] равна скорости обратной реакции: v–1 = k – 1 [ С ] • [ D ] , т. е. v+1 = v–1 соответственно k+1[А]•[B] = k–1[С]•[D], или 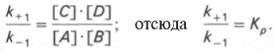 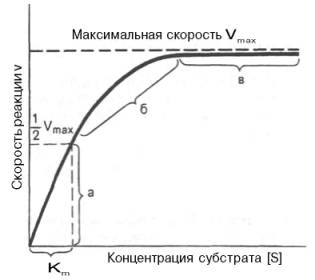 Рис. 4.12. Теоретический график зависимости скорости ферментативной реакции от концентрации субстрата при постоянной концентрации фермента. а - реакция первого порядка (при [ S ] < Кm скорость реакции пропорциональна концентрации субстрата); б - реакциясмешанного порядка; в - реакция нулевого порядка, когда v = Vmaxи скорость реакции не зависит от концентрациисубстрата. Таким образом, константа равновесия равна отношению констант скоростей прямой и обратной реакций. Величину, обратную константе равновесия, принято называть субстратной константой, или, в случае ферментативной реакции, константой диссоциации фермент–субстратного комплекса, и обозначать символом KS. Так, в реакции 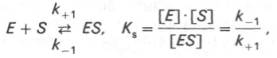 т.е. KSравна отношению произведения концентрации фермента и субстрата к концентрации фермент-субстратного комплекса или отношению констант скоростей обратной и прямой реакций. Следует отметить, что константаKSзависит от химической природы субстрата и фермента и определяет степень их сродства. Чем ниже значение KS, тем выше сродство фермента к субстрату. При изучении кинетики ферментативных реакций следует учитывать одну важную особенность этих реакций (не свойственную обычным химическим реакциям), связанную с явлением насыщения фермента субстратом. При низкой концентрации субстрата зависимость скорости реакции от концентрации субстрата (рис. 4.12) является почти линейной и подчиняется кинетике первого порядка. Это означает, что скорость реакции S —> Р прямо пропорциональна концентрации субстрата S и в любой момент времени t определяется следующим кинетическим уравнением: 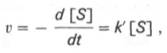 где [S] – молярная концентрация субстрата S; –d[S]/dt – скорость убыли субстрата; k' – константа скорости реакции, которая в данном случае имеет размерность, обратную единице времени (мин–1 или с–1). При высокой концентрации субстрата скорость реакции максимальна, становится постоянной и не зависящей отконцентрации субстрата [ S ] . В этом случае реакция подчиняется кинетике нулевого порядка v = k" (при полном насыщении фермента субстратом) и целиком определяется концентрацией фермента. Различают, кроме того, реакции второго порядка, скорость которых пропорциональна произведению концентраций двух реагирующих веществ. В определенных условиях при нарушении пропорциональности говорят иногда о реакциях смешанного порядка (см. рис. 4.12). Изучая явление насыщения, Л. Михаэлис и М. Ментен разработали общую теорию ферментативной кинетики. Они исходили из предположения, что ферментативный процесс протекает в виде следующей химической реакции: 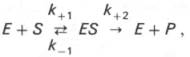 т.е. фермент Е вступает во взаимодействие с субстратом S с образованием промежуточного комплекса ES, который далее распадается на свободный фермент и продукт реакции Р. Математическая обработка на основе закона действующих масс дала возможность вывести уравнение, названное в честь авторов уравнением Михаэлиса–Ментен, выражающее количественное соотношение между концентрацией субстрата и скоростью ферментативной реакции: где v – наблюдаемая скорость реакции при данной концентрации субстрата [S]; KS– константа диссоциации фермент-субстратного комплекса, моль/л; Vmax– максимальная скорость реакции при полном насыщении ферментасубстратом. Из уравнения Михаэлиса–Ментен следует, что при высокой концентрации субстрата и низком значении KSскорость реакции является максимальной, т.е. v = Vmax(реакция нулевого порядка, см. рис. 4.12). При низкой концентрациисубстрата, напротив, скорость реакции оказывается пропорциональной концентрации субстрата в каждый данный момент (реакция первого порядка). Следует указать, что уравнение Михаэлиса–Ментен в его классическом виде не учитывает влияние на скорость ферментативного процесса продуктов реакции, например в реакции 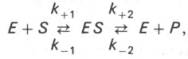 и носит несколько ограниченный характер. Поэтому были предприняты попытки усовершенствовать его. Так, было предложено уравнение Бриггса-Холдейна: где Кm представляет собой константу Михаэлиса, являющуюся экспериментально определяемой величиной. Она может быть представлена следующим уравнением: 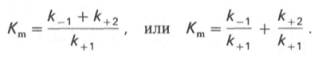 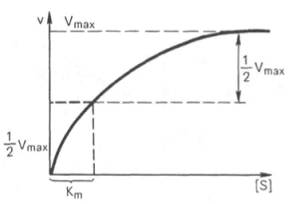 Рис. 4.13. Кривая уравнения Михаэли-са-Ментен: гиперболическая зависимость начальных скоростей катализируемой ферментом реакции от концентрации субстрата. В числителе представлены константы скоростей распада комплекса ES в двух направлениях (в сторону исходных Е и S и в сторону конечных продуктов реакции Е и Р). Отношение k–1/ k+1представляет собой константу диссоциацииферментсубстратного комплекса KS, тогда: 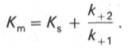 Отсюда вытекает важное следствие: константа Михаэлиса всегда больше константы диссоциации фермент-субстратного комплекса KSна величину k+2/k+1. Для определения численного значения Кm обычно находят ту концентрацию субстрата, при которой скорость ферментативной реакции v составляет половину от максимальной Vmax, т.е. если v = 1/2 Vmaх. Подставляя значение v в уравнение Бриггса–Холдейна, получаем: 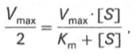 разделив обе части уравнения на Vmах, получим 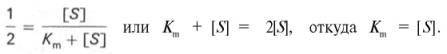 Таким образом, константа Михаэлиса численно равна концентрации субстрата (моль/л), при которой скорость данной ферментативной реакции составляет половину от максимальной. Определение величины Кm имеет важное значение при выяснении механизма действия эффекторов на активность ферментов и т.д. Константу Михаэлиса можно вычислить по графику (рис. 4.13). Отрезок на абсциссе, соответствующий скорости, равной половине максимальной, будет представлять собой Кm. Пользоваться графиком, построенным в прямых координатах зависимости начальной скорости реакции v0 от начальной концентрации субстрата [S0], неудобно, поскольку максимальная скорость Vmaxявляется в данном случае асимптотической величиной и определяется недостаточно точно. 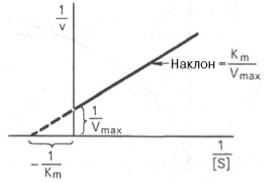 Рис. 4.14. График Лайнуивера-Бэрка. Для более удобного графического представления экспериментальных данных Г. Лайнуивер и Д. Бэрк преобразовали уравнение Бриггса–Хол-дейна по методу двойных обратных величин исходя из того принципа, что если существует равенство между двумя какими-либо величинами, то и обратные величины также будут равны. В частности, если 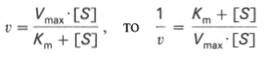 или 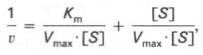 то после преобразования получаем уравнение: 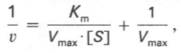 которое получило название уравнения Лайнуивера–Бэрка. Это уравнение прямой линии: у = ах + b. Если теперь в соответствии с этим уравнением построить график в координатах 1/v (y) от l/[S] (x), то получим прямую линию (рис. 4.14), тангенс угла наклона который будет равен величине Km/Vmax; отрезок, отсекаемый прямой от оси ординат, представляет собой l/Vmax(обратная величина максимальной скорости). Если продолжить прямую линию за ось ординат, тогда на абсциссе отсекается отрезок, соответствующий обратной величине константы Михаэлиса – 1/Кm (см. рис. 4.14). Таким образом, величину Кm можно вычислить из данных наклона прямой и длины отрезка, отсекаемого от оси ординат, или из длины отрезка, отсекаемого от оси абсцисс в области отрицательных значений. Следует подчеркнуть, что значения Vmax, как и величину Кm, более точно, чем по графику, построенному в прямых координатах, можно определить по графику, построенному по методу двойных обратных величин. Поэтому данный метод нашел широкое применение в современной энзимологии. Предложены также аналогичные графические способы определения Кm и Vmaxв координатах зависимости v от v/[S] и [S]/v от [S]. Следует отметить некоторые ограничения применения уравнения Ми-хаэлиса–Ментен, обусловленные множественными формами ферментов и аллостерической природой фермента. В этом случае график зависимости начальной скорости реакции от концентрации субстрата (кинетическая 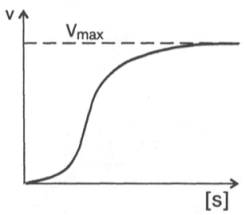 Рис. 4.15. Сигмоидная кинетическая кривая насыщения субстратом. кривая) имеет не гиперболическую форму, а сигмоидный характер (рис. 4.15) наподобие кривой насыщения гемоглобина кислородом. Это означает, что связывание одной молекулы субстрата в одном каталитическом центре повышает связывание субстрата с другим центром, т.е. имеет место кооперативное взаимодействие, как и в случае присоединения кислорода к 4 субъединицам гемоглобина. Для оценки концентрации субстрата, при которой скорость реакции составляет половину максимальной, в условиях сигмоидного характера кинетической кривой обычно применяют преобразованное уравнение Хилла: 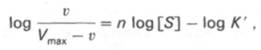 где К' – константа ассоциации; n – число субстратсвязывающих центров. 11. Назовите факторы, которые влияют на скорость ферментативной реакции. Охарактеризуйте влияние этих факторов на скорость ферментативной реакции. Одним из наиболее существенных факторов, определяющих скорость ферментативной реакции, является концентрация субстрата (или субстратов) и продукта (продуктов). При постоянной концентрации фермента скорость реакции постепенно увеличивается, достигая определенного максимума, когда дальнейшее увеличение количества субстрата практически не оказывает влияния на скорость ферментативной реакции. В таких случаях принято считать, что субстрат находится в избытке, а фермент полностью насыщен, т.е. все молекулы фермента связаны с субстратом. Ограничивающим скорость реакции фактором в последнем случае становится концентрация фермента. Именно при этих условиях определяют величину максимальной скорости (Vmax) и значения константы Михаэлиса (Km) (см. рис. 4.13; 4.14). Скорость любой ферментативной реакции непосредственно зависит от концентрации фермента. Существующая линейная зависимость между этими величинами, когда скорость реакции прямо пропорциональна количеству присутствующего фермента, справедлива только в определенных условиях, например в начальный период ферментативной реакции, так как в этот период практически не происходит обратной реакции, а концентрация продукта оказывается недостаточной для обратимости реакции. Именно в этом случае скорость реакции (точнее, начальная скорость реакции v) будет пропорциональна концентрации фермента. Как было отмечено, фермент является одной из реагирующих молекул в химической реакции и при взаимодействии с субстратом образует промежуточный фермент-субстратный комплекс, который далее подвергается распаду на продукт и свободный фермент: 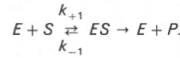 Если упростить это уравнение, исключив промежуточный ES-комплекс: 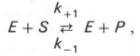 то в уравнениях для скоростей прямой и обратной реакций обязательным компонентом является концентрация фермента: Однако в уравнениях для константы равновесия (Keqили Кр) концентрация фермента уже не имеет значения: 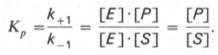 Как видно, константа равновесия (Kр) ферментативной реакции не зависит от концентрации фермента. Определяя скорость и направление химической реакции, фермент тем не менее не оказывает влияния на конечные (равновесные) концентрации реагирующих молекул и продуктов, определяющих величину константы равновесия. 12. Регуляция активности ферментов; пути ее активации и инактивации. Влияние ионизирующего излучения и экологических факторов на ферменты. Одним из уникальных свойств живых организмов является удивительная их способность к сохранению сбалансированности катаболических (биодегра-дативных) и анаболических (биосинтетических) процессов. При этом в клетках одновременно совершаются процессы синтеза, распада и взаимопревращения сотен и тысяч разнообразных веществ, которые в свою очередь регулируются множеством механизмов, обеспечивающих постоянство внутренней среды организма. Некоторые из этих регуляторных механизмов, среди которых важная роль принадлежит механизмам регуляции синтеза и каталитической активности ферментов, будут рассмотрены далее. Влияние закона действия масс. В катализируемой ферментом обратимой химической реакции, например А + В <=> С + D, концентрация компонентов реакции и соответственно направление реакции будут регулироваться влиянием закона действия масс. Оно, в частности, может быть показано в обратимой реакции трансаминирования, катализируемой ферментом аланинаминотрансферазой: Аланин + α-Кетоглутарат <=> Пируват + Глутамат. Этот тип регуляции играет, очевидно, лишь ограниченную роль, поскольку в реальных условиях реакция обычно протекает в одном направлении, так как образовавшиеся продукты могут оказаться субстратами для действия других ферментов и выводиться из сферы реакции. В этих случаях устанавливается скорее устойчивое (стационарное) состояние, чем истинное равновесие. Изменение количества фермента. На бактериях хорошо изучен феномен индуцированного (индуцирующего) синтеза ферментов при выращивании их на среде, где единственным источником углерода и энергии служит тот или иной углевод, например глюкоза. Замена в среде глюкозы на лактозу (индуктор) приводит к индуцированному или адаптивному (после небольшого периода лаг-фазы) синтезу фермента галактозидазы (программированному лактозным геном, см. главу 13), расщепляющей лактозу на глюкозу и галактозу. В клетках прокариот и эукариот имеются ферменты, концентрация которых не требует добавления индуктора; это так называемые конститутивные ферменты. Количество фермента в клетке зависит от наличия продукта реакции, катализируемой данным ферментом, причем продукт реакции вызывает торможение синтеза фермента в результате репрессии (см. далее). В животных тканях быстрый синтез ферментов наблюдается реже. Механизм его (индуцирующий синтез) изучен только для небольшого числа ферментов: тирозинтрансаминазы, серин- и треониндегидратазы, триптофанпирролазы и др. – в ответ на введение гормонов или прием белковой пищи. Однако при поступлении в организм некоторых ядов, канцерогенных веществ, алкалоидов, инсектицидов через несколько дней наблюдается резкое повышение активности(соответственно количества) ферментов-гидроксилаз (монооксигеназ) эндоплазматической сети клеток печени, окисляющих чужеродные вещества в нетоксичные для организма продукты. Вполне допустимо предположить, что в этих случаях имеет место синтез ферментов путем индукции (т.е. de novo). Описаны случаи, когда под действием подобных гидроксилаз чужеродные вещества превращаются в организме в более токсичные соединения. Это явление, обратное детоксикации, получило название летального синтеза. Проферменты. Протеолитические ферменты пищеварительного тракта, а также поджелудочной железы синтезируются в неактивной форме – в виде проферментов (зимогенов). Регуляция в этих случаях сводится к превращению проферментов в активные ферменты под влиянием специфических агентов или других ферментов – протеиназ. Так, трипсин в поджелудочной железе синтезируется в форме неактивного трипсиногена. Поступив в кишечник, он превращается в активный трипсин в результате аутокатализа или под действием других протеиназ (механизм активации подробно рассматривается в главе 12). Превращение неактивного пепси-ногена в активный пепсин происходит аутокаталитически в результате специфического ограниченного протеолиза в присутствии соляной кислоты и также связано с отщеплением от профермента специфического ингибитора пептидной природы. Эти превращения зимогенов в активные ферменты связаны с конформационными изменениями молекулы фермента и формированием активного центра или его раскрытием (демаскирование). Синтез протеиназ в неактивной форме и ряда других неактивных белков-предшественников имеет, очевидно, определенный биологический смысл, предотвращая разрушение клеток органов, в которых образуются проферменты. Примерами подобного активирования белков является активирование некоторых гормонов (проинсулин —> инсулин), белка соединительной ткани (растворимый проколлаген превращается в нерастворимый коллаген), белков свертывающей системы крови.  Рис. 4.23. Ковалентная модификация фермента путем фосфорилирования-дефосфо-рилирования остатков серина.  Рис. 4.24. Нековалентная модификация фермента путем аденилирования-деадени-лирования. Химическая модификация фермента. Некоторые белки при формировании третичной структуры подвергаются постсинтетической химической модификации (см. главу 1). Оказалось, что активность ряда ключевых ферментовобмена углеводов, в частности фосфорилазы, гликогенсинтазы и др., также контролируется путем фосфорилирования и дефосфорили-рования, осуществляемого специфическими ферментами – протеинкиназой и протеинфосфатазой, активность которых в свою очередь регулируется гормонами (см. главу 10). Уровень активностиключевых ферментов обмена углеводов и соответственно интенсивность и направленность самих процессов обмена определяются соотношением фосфорилированных и де-фосфорилированных форм этих ферментов. Обычно различают обратимую ковалентную и нековалентную химические модификации ферментов, осуществляемые через ОН-группы серина, реже – тирозина или за счет нековалентных взаимодействий с молекулой фермента. В первом случае активным ферментом оказывается или фосфо-рилированная, или дефосфорилированная форма, как в случае с молекулами мышечной фосфорилазы и гликогенсинтазы соответственно (см. главу 10). В качестве примеров можно в виде схемы представить оба типа модификации, в которой символом Р обозначается остаток фосфата, Pi– неорганический фосфат (Н3РО4), РРi – неорганический пирофосфат (Н4Р2О7), АМФ – остаток адениловой кислоты (рис. 4.23; 4.24). Химическая постсинтетическая модификация ферментов включает, кроме того, процессы ограниченного протеолиза(см. ранее), метилирования (см. главу 13), гликозилирования, уридилирования, аденилирования, АДФ-рибозилирования и др., обеспечивая тем самым микроскопический тип регуляции активности ферментов и соответственно физиологическую скорость процессов обмена веществ. Аллостерическая регуляция. Во многих строго биосинтетических реакциях основным типом регуляции скорости многоступенчатого ферментативного процесса является ингибирование по принципу обратной связи. Это означает, что конечный продукт биосинтетической цепи подавляет активность фермента, катализирующего первую стадию синтеза, которая является ключевой для данной цепи реакции. Поскольку конечный продукт структурно отличается от субстрата, он связывается с аллостери-ческим (некаталитическим) центром молекулы фермента, вызывая ингиби-рование всей цепи синтетической реакции. Предположим, что в клетках осуществляется многоступенчатый биосинтетический процесс, каждая стадия которого катализируется собственным ферментом: Скорость подобной суммарной последовательности реакций в значительной степени определяется концентрациейконечного продукта Р, накопление которого выше допустимого уровня оказывает мощное инги-бирующее действие на первую стадию процесса и соответственно на фермент E1. Впервые существование подобного механизма контроля активности ферментов метаболитами было обнаружено у Е.coli при исследовании синтеза изолейцина и ЦТФ. Оказалось, что изолейцин, являющийся конечным продуктом синтеза, избирательно подавляет активность треонин-дегидратазы, катализирующей первую стадию последовательного процесса превращения треонина в изолейцин, насчитывающего пять ферментативных реакций:  Аналогично ЦТФ как конечный продукт биосинтетического пути оказывает ингибирующий эффект на первый фермент(аспартаткарбамоилтран-сферазу), регулируя тем самым свой собственный синтез (см. главу 13). Этот тип ингибирования получил название ингибирования по принципу обратной связи, или ретроингибирования. Существование его доказано во всех живых организмах. В настоящее время он рассматривается как один из ведущих типов регуляции активности ферментов и клеточного метаболизма в целом. 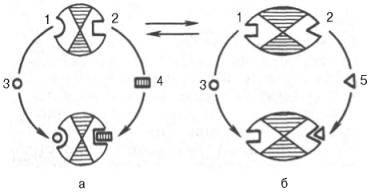 Рис. 4.25. Взаимодействие алло-стерического фермента с субстратом и эффекторами (схема). а - активный комплекс; б - неактивный комплекс; 1 - активный центр; 2 - аллостерический центр; 3 - субстрат; 4 - положительный эффектор; 5 - отрицательный эффектор. С другой стороны, в амфиболических процессах, выполняющих одновременно биосинтетические и биодеградативные функции , доказано существование регуляции как по типу ретроингибирования, так и макроэрги-ческими соединениями – индикаторами энергетического состояния клетки. Для амфиболических процессов уникальным типом регуляции, свойственным только им, является, кроме того, активация предшественником, когда первый метаболит в многоступенчатом пути активирует фермент, катализирующий последнюю стадию. Так, доказано активирующее влияние глюкозо-6-фосфата, являющегося предшественником гликогена, на фермент гликогенсинтазу. Подобные типы ингибирования конечным продуктом и активирования первым продуктом свойственны аллостерическим (регуляторным) ферментам, когда эффектор, модулятор, структурно отличаясь от субстрата, связывается в особом (аллостерическом) центре молекулы фермента, пространственно удаленном от активного центра. Следует, однако, иметь в виду, что модуляторами аллостерических ферментов могут быть как активаторы, так и ингибиторы. Часто оказывается, что сам субстрат оказывает активирующий эффект. Ферменты, для которых и субстрат, и модулятор представлены идентичными структурами, носят название гомотропных в отличие от гетеротропных ферментов, для которых модулятор имеет отличную от субстрата структуру. Взаимопревращение активного и неактивного аллостерических ферментов в упрощенной форме, а также конфор-мационные изменения, наблюдаемые при присоединении субстрата и эффекторов, представлены на рис. 4.25. Присоединение отрицательного эффектора к аллостерическому центру вызывает значительные изменения конфигурации активного центра молекулы фермента, в результате чего фермент теряет сродство к своему субстрату (образование неактивного комплекса). Аллостерические взаимодействия проявляются в характере кривых зависимости начальной скорости реакции от концентрации субстрата или эффектора, в частности в S-образности этих кривых (отклонение от гиперболической кривой Михаэлиса-Ментен). S-образный характер зависимости v от [ S ] в присутствии модулятора обусловлен эффектом кооперативности. Это означает, что связывание одной молекулы субстрата облегчает связывание второй молекулы в активном центре, способствуя тем самым увеличению скорости реакции. Кроме того, для аллостерических регуляторных ферментов характерна нелинейная зависимость скорости реакции от концентрациисубстрата. Другие типы регуляции активности ферментов. Абсолютное количество присутствующего в клетке фермента регулируется временем его синтеза и распада. К регуляторным механизмам могут быть отнесены также конкуренция ферментов за общий субстрат, выключение активности одного из изоферментов (у множественных форм ферментов), влияние концентраций кофакторов и явление компартментализации. Механизм компарт-ментализации метаболических процессов играет, по-видимому, важную биологическую роль, пространственно разъединяя с помощью биомембран ферментысо своими субстратами (например, лизосомальные ферменты: протеиназы, фосфатазы, рибонуклеазы и другие гидролитические ферменты – с цитоплазматическими веществами, на которые они действуют). Кроме того, облегчая независимую регуляцию, этот механизм позволяет разделить несовместимые в одном и том же месте (и, возможно, в одно и то же время) метаболические процессы. Примером последних могут быть пути синтеза высших жирных кислот, протекающие в основном в растворимой фракции цитоплазмы, и пути распада (окисления) жирных кислот, сосредоточенные в митохондриях. Необходимо указать, однако, что при ком-партментализации возникает проблема транспорта как метаболитов, так и восстановительных эквивалентов через биомембраны субклеточных ор-ганелл. Эту задачу решает так называемый челночный механизм, позволяющий перевод метаболитов в формы, способные переходить через мембраны, и обеспечивающий внутриклеточный гомеостаз (см. главу 13). 13. Молекулярные механизмы регуляции активности ферментов. Активность ферментов в клетке непостоянна во времени. Ферменты чутко реагируют на ситуацию, в которой оказывается клетка, на факторы, воздействующие на нее как снаружи, так и изнутри. Главная цель такой чувствительности ферментов – отреагировать на изменение окружающей среды, приспособить клетку к новым условиям, дать должный ответ на гормональные и иные стимулы, а в некоторых ситуациях – предоставить клетке шанс выжить. |