Значение биохимии в подготовке врача. Биологическая химия
 Скачать 8.33 Mb. Скачать 8.33 Mb.
|

КоА + АМФ + PPi. |
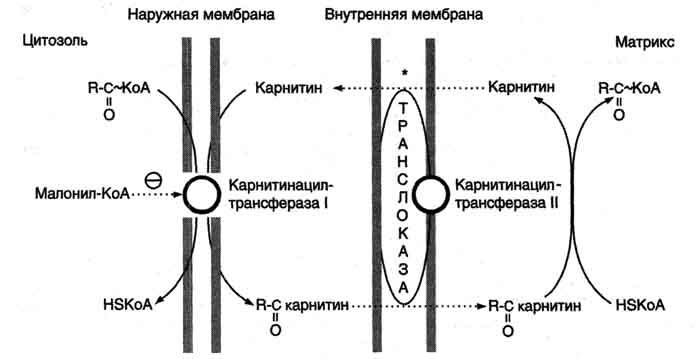
| β-Окисление | Кол-во АТФ |
| 7 NADH (от пальмитоил-КоА до ацетил-КоА), окисление каждой молекулы кофермента в ЦПЭ обеспечивает синтез 3 молекул АТФ | 21 |
| 7 FADHa, окисление каждой молекулы кофермента в ЦПЭ обеспечивает синтез 2 молекул АТФ | 14 |
| Окисление каждой из 8 молекул ацетил-КоА в ЦТК обеспечивает синтез 12 молекул АТФ | 96 |
| Сумм. кол-во молекул АТФ, синтезированных при окислении одной молекулы пальмитоил-КоА | 131 |
Регуляция скорости β-окисления
β-Окисление - метаболический путь, прочно связанный с работой ЦПЭ и общего пути катаболизма. Поэтому его скорость регулируется потребностью клетки в энергии, т.е. соотношениями АТФ/АДФ и NADH/NAD+, так же, как и скорость реакций ЦПЭ и общего пути катаболизма (см. раздел 6). Скорость β-окисления в тканях зависит от доступности субстрата, т.е. от количества жирных кислот, поступающих в митохондрии. Концентрация свободных жирных кислот в крови повышается при активации ли-полиза в жировой ткани при голодании под действием глюкагона и при физической работе под действием адреналина. В этих условиях жирные кислоты становятся преимущественным источником энергии для мышц и печени, так как в результате β-окисления образуются NADH и ацетил-КоА, ингибирующие пируватдегидрогеназный комплекс. Превращение пирувата, образующегося из глюкозы, в ацетил-КоА замедляется. Накапливаются промежуточные метаболиты гликолиза и, в частности, глюкозо-6-фосфат. Глюкозо-6-фосфат ингибирует гексокиназу и, следовательно, препятствует использованию глюкозы в процессе гликолиза. Таким образом, преимущественное использование жирных кислот как основного источника энергии в мышечной ткани и печени сберегает глюкозу для нервной ткани и эритроцитов.
Скорость β-окисления зависит также от активности фермента карнитинацилтрансферазы I. В печени этот фермент ингибируется малонил-КоА, веществом, образующимся при биосинтезе жирных кислот. В абсорбтивный период в печени активируется гликолиз и увеличивается образование ацетил-КоА из пирувата. Первая реакция синтеза жирных кислот - превращение ацетил-КоА в малонил-КоА. Малонил-КоА ингибирует β-окисление жирных кислот, которые могут использоваться для синтеза жира.
Окисление ненасыщенных жирных кислот
Около половины жирных кислот в организме человека ненасыщенные. β-Окисление этих кислот идёт обычным путём до тех пор, пока двойная связь не окажется между третьим и четвёртым атомами углерода (рис. 8-28). Затем фермент еноил-КоА изомераза перемещает двойную связь из положения 3-4 в положение 2-3 и изменяет цис-конформацию двойной связи на транс-, которая требуется для р-окисления. В этом цикле Р-окисления первая реакция дегидрирования не происходит, так как двойная связь в радикале жирной кислоты уже имеется. Далее циклы β-окисления продолжаются, не отличаясь от обычного пути.
α-Окисление жирных кислот. В липидах мозга и других отделах нервной ткани преобладают жирные кислоты с очень длинной цепью - более 20 углеродных атомов. Они окисляются по типу α-окисления, при котором от жирной кислоты отщепляется по одному атому углерода, выделяющемуся в виде СО2 (рис. 8-29).
Этот путь катаболизма жирных кислот не связан с синтезом АТФ. α-Окислению подвергаются также жирные кислоты с разветвлённой углеводородной цепью, например фитановая, поступающая в организм с растительной пищей (рис. 8-30). Фитановая кислота образуется из фитола, который входит в состав хлорофилла. В этой кислоте у каждого третьего атома углерода находится метильная группа, что делает невозможным β-окисление данной кислоты. При α-окислении фитановой кислоты вначале удаляется метильная группа, а затем происходит цикл р-окисления.
67. Биосинтез жирных кислот. Основные стадии процесса. Регуляция обмена жирных кислот.
Синтез жиров в жировой ткани и печени
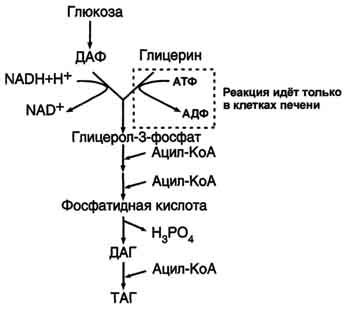
Синтез жиров происходит в абсорбтивный период в печени и жировой ткани. Непосредственными субстратами в синтезе жиров являются ацил-КоА и глицерол-3-фосфат. Метаболический путь синтеза жиров в печени и жировой ткани одинаков, за исключением разных путей образования глицерол-3-фосфата.
Образование глицерол-3-фосфата
Синтез жиров в печени и жировой ткани идёт через образование промежуточного продукта - фосфатидной кислоты (рис. 8-21).
Предшественник фосфатидной кислоты - глицерол-3-фосфат, образующийся в печени двумя путями:
восстановлением дигидроксиацетонфосфата - промежуточного метаболита гликолиза;
фосфорилированием глицеролкиназой свободного глицерола, поступающего в печень из крови (продукт действия ЛП-липазы на жиры ХМ и ЛПОНП).
В жировой ткани глицеролкиназа отсутствует, и восстановление дигидроксиацетонфосфата - единственный путь образования глицерол-3-фосфата. Следовательно, синтез жиров в жировой ткани может происходить только в абсорбтивный период, когда глюкоза поступает в адипоциты с помощью белка-переносчика глюкозы ГЛЮТ-4, активного только в присутствии инсулина, и распадается по пути гликолиза.
Синтез жиров в жировой ткани
В жировой ткани для синтеза жиров используются в основном жирные кислоты, освободившиеся при гидролизе жиров ХМ и ЛПОНП (рис. 8-22). Жирные кислоты поступают в адипоциты, превращаются в производные КоА и взаимодействуют с глицерол-3-фосфатом, образуя сначала лизофосфатидную кислоту, а затем фосфатидную. Фосфатидная кислота после дефосфорилирования превращается в диацилглицерол, который ацилируется с образованием триацилглицерола.
Кроме жирных кислот, поступающих в адипоциты из крови, в этих клетках идёт и синтез жирных кислот из продуктов распада глюкозы. В адипоцитах для обеспечения реакций синтеза жира распад глюкозы идёт по двум путям: гликолиз, обеспечивающий образование глицерол-3-фосфата и ацетил-КоА, и пентозофосфатный путь, окислительные реакции которого обеспечивают образование NADPH, служащего донором водорода в реакциях синтеза жирных кислот.
Молекулы жиров в адипоцитах объединяются в крупные жировые капли, не содержащие воды, и поэтому являются наиболее компактной формой хранения топливных молекул. Подсчитано, что, если бы энергия, запасаемая в жирах, хранилась в форме сильно гидратированных молекул гликогена, то масса тела человека увеличилась бы на 14-15 кг.
Синтез ТАГ в печени. Образование ЛПОНП в печени и транспорт жиров в другие ткани
Печень - основной орган, где идёт синтез жирных кислот из продуктов гликолиза. В гладком ЭР гепатоцитов жирные кислоты активируются и сразу же используются для синтеза жиров, взаимодействуя с глицерол-3-фосфатом. Как и в жировой ткани, синтез жиров идёт через образование фосфатидной кислоты. Синтезированные в печени жиры упаковываются в ЛПОНП и сек-ретируются в кровь.
В состав ЛПОНП, кроме жиров, входят холестерол, фосфолипиды и белок - апоВ-100. Это очень "длинный" белок, содержащий 11 536 аминокислот. Одна молекула апоВ-100 покрывает поверхность всего липопротеина.
ЛПОНП из печени секретируются в кровь, где на них, как и на ХМ, действует ЛП-липаза. Жирные кислоты поступают в ткани, в частности в адипоциты, и используются для синтеза жиров. В процессе удаления жиров из ЛПОНП под действием ЛП-липазы ЛПОНП сначала превращаются в ЛГШП, а затем в ЛПНП. В ЛПНП основными липидными компонентами служат холестерол и его эфиры, поэтому ЛПНП являются липопротеинами, доставляющими холестерол в периферические ткани. Глицерол, освободившийся из липопротеинов, кровью транспортируется в печень, где опять может использоваться для синтеза жиров.
Скорость синтеза жирных кислот и жиров в печени существенно зависит от состава пищи. Если в пище содержится более 10% жиров, то скорость синтеза жиров в печени резко снижается.
Лимитирующим ферментом является ацетил-КоАкарбоксилаза. Аллостерические активаторы — АТФ и цитрат, ингибиторы — жирные кислоты с длинной цепью. Инсулин, эстрогены активиру¬ют, катехоламины и стресс ингибируют синтез жирных кислот. Значение:при распаде УВ обр ацетил-Коа, который используется в синтезе ЖК, т.о. избыток УВ запасается в виде жира.
68. Кетоновые тела, биосинтез и использование в качестве источников энергии. Причины развития кетонемии и кетонурии при голодании и сахарном диабете. Склонность к кетозу у детей.
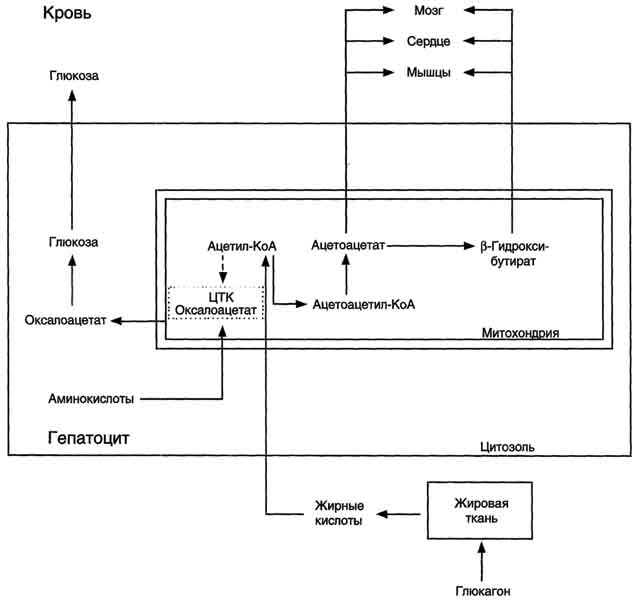
Рис. 8-32. Активация синтеза кетоновых тел при голодании. Точечные линии - скорость метаболических путей снижена; сплошные линии - скорость метаболических путей повышена. При голодании в результате действия глюкагона активируются липолиз в жировой ткани и (3-окисление в печени. Количество оксалоацетата в митохондриях уменьшается, так как он, восстановившись до малата, выходит в цитозоль, где опять превращается в Оксалоацетат и используется в глюконеогенезе. В результате скорость реакций ЦТК снижается и, соответственно, замедляется окисление ацетил-КоА. Концентрация ацетил-КоА в митохондриях увеличивается, и активируется синтез кетоновых тел. Синтез кетоновых тел увеличивается также при сахарном диабете.
К кетоновым телам относят β-гидроксибутират, ацетоацетат и ацетон. Первые две молекулы могут окисляться в тканях, обеспечивая синтез АТФ. Ацетон образуется только при высоких концентрациях кетоновых тел в крови и, выделяясь с мочой, выдыхаемым воздухом и потом, позволяет организму избавляться от избытка кетоновых тел.
Синтез кетоновых тел в печени. При низком соотношении инсулин/глюкагон в крови в жировой ткани активируется распад жиров. Жирные кислоты поступают в печень в большем количестве, чем в норме, поэтому увеличивается скорость β-окисления. Скорость реакций ЦТК в этих условиях снижена, так как оксалоацетат используется для глюконеогенеза. В результате скорость образования ацетил-КоА превышает способность ЦТК окислять его. Ацетил-КоА накапливается в митохондриях печени и используется для синтеза кетоновых тел. Синтез кетоновых тел происходит только в митохондриях печени.
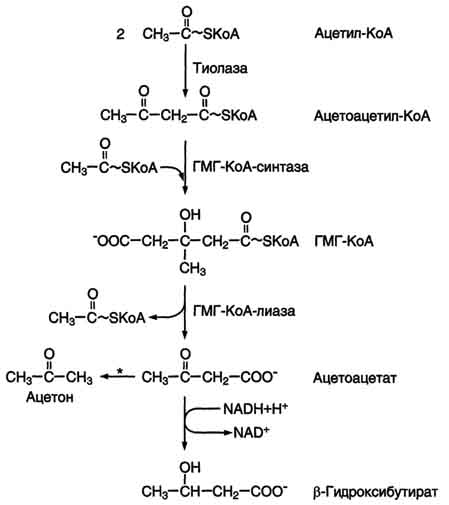
Синтез кетоновых тел начинается с взаимодействия двух молекул ацетил-КоА, которые под действием фермента тиолазы образуют ацетоацетил-КоА. С ацетоацетил-КоА взаимодействует третья молекула ацетил-КоА, образуя 3-гидрокси-3-метилглутарил-КоА (ГМГ-КоА). Эту реакцию катализирует фермент ГМГ-КоА-синтаза. Далее ГМГ-КоА-лиаза катализирует расщепление ГМГ-КоА на свободный ацетоацетат и ацетил-КоА.
Ацетоацетат может выделяться в кровь или превращаться в печени в другое кетоновое тело - β-гидроксибутират путём восстановления.
В клетках печени при активном β-окислении создаётся высокая концентрация NADH. Это способствует превращению большей части ацетоацетата в β-гидроксибутират, поэтому основное кетоновое тело в крови - именно β-гидроксибутират. При голодании для многих тканей жирные кислоты и кетоновые тела становятся основными топливными молекулами. Глюкоза используется в первую очередь нервной тканью и эритроцитами.
При высокой концентрации ацетоацетата часть его неферментативно декарбоксилируется, превращаясь в ацетон. Ацетон не утилизируется тканями, но выделяется с выдыхаемым воздухом и мочой. Таким путём организм удаляет избыточное количество кетоновых тел, которые не успевают окисляться, но, являясь водорастворимыми кислотами, вызывают ацидоз.
Рис. 8-33. Синтез кетоновых тел в митохондриях гепатоцитов. Регуляторный фермент синтеза кетоновых тел (ГМГ-КоА-синтаза) ингибируется свободным КоА. - реакция идёт неферментативно при высокой концентрации кетоновых тел в крови.
Окисление кетоновых тел в периферических тканях
При длительном голодании кетоновые тела становятся основным источником энергии для скелетных мышц, сердца и почек. Таким образом глюкоза сохраняется для окисления в мозге и эритроцитах. Уже через 2-3 дня после начала голодания концентрация кетоновых тел в крови достаточна для того, чтобы они проходили в клетки мозга и окислялись, снижая его потребности в глюкозе.
β-Гидроксибутират, попадая в клетки, дегидрируется NAD-зависимой дегидрогеназой и превращается в ацетоацетат. Ацетоацетат активируется, взаимодействуя с сукцинил-КоА - донором КоА:
Ацетоацетат + Сукцинил-КоА → Ацетоацетил- КоА + Сукцинат.
У детей до 7 лет отмечается повышенная склонность к кетозам. Они могут развиваться под влиянием различных причин: кратковременное голодание, переутомление, перевозбуждение, инфекционные заболевания. Вследствие неустойчивости углеводного обмена и малых запасов гликогена удовлетворение энергозатрат происходит за счет интенсивного окисления СЖК. Из-за несовершенства выделительной системы почек, образующиеся при этом кетоновые тела накапливаются в крови, вызывая приступы неукротимой рвоты «ацетонемическая рвота»
69. Холестерин. Пути поступления, использования и выведения из организма. Уровень холестерина в сыворотке крови. Биосинтез холестерина, его этапы. Регуляция синтеза.
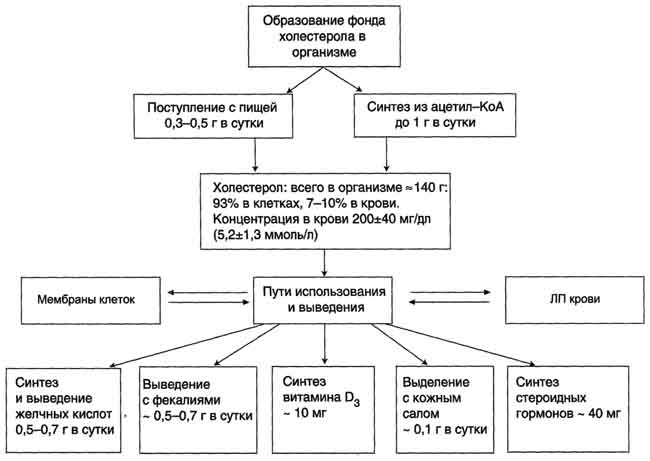
Холестерол - стероид, характерный только для животных организмов. Он синтезируется во многих тканях человека, но основное место синтеза - печень. В сутки в организме синтезируется около 1 г холестерола; с пищей поступает 300-500 мг. Холестерол выполняет много функций: входит в состав всех мембран клеток и влияет на их свойства, служит исходным субстратом в синтезе жёлчных кислот и стероидных гормонов. Предшественники в метаболическом пути синтеза холестерола превращаются также в убихинон - компонент дыхательной цепи и долихол, участвующий в синтезе гликопротеинов. Холестерол за счёт своей гидроксильной группы может образовывать эфиры с жирными кислотами. Этерифицированный холестерол преобладает в крови и запасается в небольших количествах в некоторых типах клеток, использующих его как субстрат для синтеза других веществ. Холестерол и его эфиры - гидрофобные молекулы, поэтому они транспортируются кровью только в составе разных типов ЛП. Обмен холестерола чрезвычайно сложен - только для его синтеза необходимо осуществление около 100 последовательных реакций. Всего в обмене холестерола участвует около 300 разных белков. Нарушения обмена холестерола приводят к одному из наиболее распространённых заболеваний - атеросклерозу. Смертность от последствий атеросклероза (инфаркт миокарда, инсульт) лидирует в общей структуре смертности населения. Атеросклероз - "полигенное заболевание", т.е. в его развитии участвуют многие факторы, важнейшие из которых наследственные. Накопление холестерола в организме приводит к развитию и другого распространённого заболевания - желчнокаменной болезни.
Реакции синтеза холестерола происходят в цитозоле клеток. Это один из самых длинных метаболических путей в организме человека.
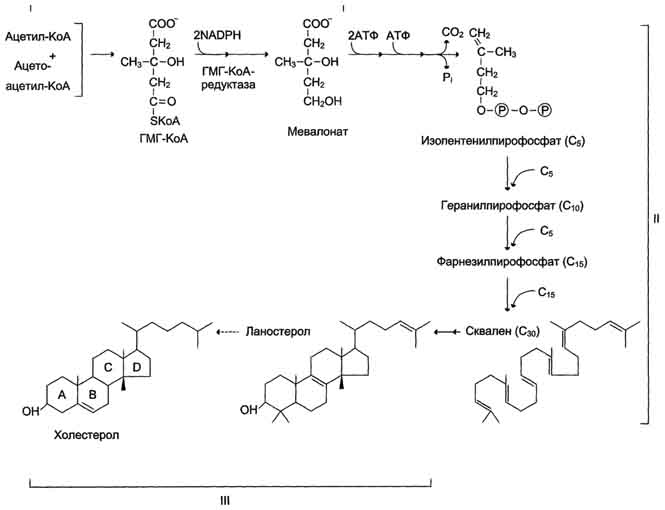
Путь синтеза холестерола можно разделить на 3 этапа. Первый этап заканчивается образованием мевалоната (мевалоновой кислоты). На втором этапе синтеза мевалонат превращается в пятиуглеродную изопреноидную структуру, содержащую пирофосфат - изопентенилпирофосфат. На третьем этапе синтеза холестерола сквален через стадию образования эпоксида ферментом циклазой превращается в молекулу ланостерола, содержащую 4 конденсированных цикла и 30 атомов углерода. Далее происходит 20 последовательных реакций, превращающих ланостерол в холестерол. На последних этапах синтеза от ланостерола отделяется 3 атома углерода, поэтому холестерол содержит 27 углеродных атомов.
Синтез холестерола. С5 - изопентенилпирофосфат; С1 - Фарнезилпирофосфат. Все атомы углерода холестерола происходят из ацетил-КоА. Сквален - углеводород линейной структуры - превращается ферментом циклазой в ланостерол, содержащий 4 конденсированных кольца и гидроксильную группу. Ланостерол через ряд последовательных реакций превращается в холестерол (I, II, III - этапы синтеза).
У холестерола имеется насыщенная разветвлённая боковая цепь из 8 углеродных атомов в положении 17, двойная связь в кольце В между атомами углерода в положениях 5 и 6, а также гидроксильная группа в положении 3.
Регуляция ключевого фермента синтеза холестерола (ГМГ-КоА-редуктазы) происходит разными способами.
Фосфорилирование/дефосфорилирование ГМГ-КоА-редуктазы. При увеличении соотношения инеулин/глюкагон этот фермент дефосфорилируется и переходит в активное состояние. Действие инсулина осуществляется через 2 фермента:
фосфатазу киназы ГМГ-КоА-редуктазы, которая превращает киназу в неактивное дефосфорилированное состояние;
фосфатазу ГМГ-КоА-редуктазы путём превращения её в дефосфорилированное активное состояние. Результатом этих реакций служит образование дефосфорилированной активной формы ГМГ-КоА-редуктазы.
Следовательно, в абсорбтивный период синтез холестерола увеличивается. В этот период увеличивается и доступность исходного субстрата для синтеза холестерола - ацетил-КоА (в результате приёма пищи, содержащей углеводы и жиры, так как ацетил-КоА образуется при распаде глюкозы и жирных кислот).
Выведение холестерола из организма
Структурная основа холестерола - кольца циклопентанпергидрофенантрена - не может быть расщеплена до СО2 и воды, как другие органические компоненты, поступающие с пищей или синтезированные в организме. Поэтому основное количество холестерола выводится в виде жёлчных кислот.
Некоторое количество жёлчных кислот выделяется в неизменённом виде, а часть подвергается действию ферментов бактерий в кишечнике. Продукты их разрушения (в основном, вторичные жёлчные кислоты) выводятся из организма.