блок_2[1]. 1. Методы синтеза предельных и ,непредельных альдегидов и кетонов из спиртов, галоидных алкилов, из карбоновых кислот и их производных, алкенов, алкинов (реакция Кучерова),
 Скачать 1.22 Mb. Скачать 1.22 Mb.
|

|
18. Органические соединения серы: тиоспирты, тиоэфиры. Со поставление их свойств со свойствами соответствующих кислород содержащих соединений.Тиоспирты или меркаптаны (хим.)Их общая формула R—SH, где R жирный или ароматический углеводородный остаток. В последнем случае тиоспирты называются тиофенолами. Эти сернистые соединения отвечают по составу спиртам, в которых водный остаток ОН замещен сульфгидрилом SH, и являются, таким образом, сложными эфирами сероводорода. Открыты Цейзе, но истинный состав их был впервые установлен Либихом. Они встречаются в природе в числе многих других продуктов гниения белковых веществ и получаются искусственно путем следующих реакций. 1) Действием пятисернистого фосфора на спирты, напр.: 5С2H5—ОН + P2S5 = 5С2H5—SH + P2O5. Эта реакция протекает нечисто и дает очень малые выходы. 2) Восстановлением хлорангидридов сульфокислот водородом в момент выделения, для чего обыкновенно берут цинк и слабую серную кислоту: С2H5SO4Cl + 3Н2 = С2H5—SH + 2Н2О + HCl. 3) Действием спиртового раствора сульфгидрата калия на галоидалкилы при нагревании, напр.: С2H5J + KSH = С2H5—SH + KJ. Вместо галоидалкилов можно взять соли серноэфирных кислот и перегонять их с избытком водного раствора сульфгидрата калия: С2H5—SO4K + KSH = С2H5—SH + K2SO4. Это самый употребительный способ получения Т. Св-ва. Серная кислота как окислитель: 2С2H5—SH + H2SO4 = (С2H5S)2 + SO2 + 2Н2О. В случае со спиртами серная кислота действует как водоотнимающее средство, в результате чего получается получ. непредельные у\в С2H5—ОH+H2SO4 =C 2H4+H2O. Более сильные окислители, напр. азотная кислота, переводят Т. в сульфокислоты:С2H5—SH + O3 = С2H5—SO2—OH. В случае когда действуешь азотной к-й на спирты они окис-ся до кислот: RCH2OH+(HNO3)=RCOOH Для положительной характеристики Т. важны их солеобразные соединения с металлами, так наз. меркаптиды, которые образуются при действии на Т. щелочей и окисей тяжелых металлов. 2С2H5—SH + HgO = (С2H5S)2Hg + H2O. В спитрах, получ-ся альдегид: C 2H 5OH+CuO=CH3-COH+H2O+ Cu Меркаптиды некоторых тяжелых металлов образуются также при действии на Т. уксуснокислых и др. солей. При нагревании большинство меркаптидов разлагается на тиоэфиры и сернистые металлы, напр.: (СН3)2Pb = PbS + (CH3)2S. Ртутные соли выделяют металл и дитиоэфир:(С2H5S)2Hg = Hg + (С2H5—S)2. 2С2H5—SH + S2Cl2 = (С2H5)2S4 + 2HCl. Тиоэфиры — соединения общей формулы (СnНm—1)2S, т. е. представляют сероводород, в котором оба водородных атома замещены углеводородными остатками (предельными и непредельными). Предельные Т. (CnH2n+1)2S получают, действуя сернистым фосфором на простые эфиры или сернистыми щелочными металлами на сложные эфиры или галоидалкилы, напр.: 2C2H5J + K2S = (C2H5)2S + 2KJ; 2C2H5O—SO2—OK + K2S = 2K2SO4 + (C2H5)2S. Смешанные Т. удобно готовить действием галоидалкилов на натриевые производные меркаптанов: C2H5SNa + CH3J = NaJ + C2H5—S—CH3 или перегонкой из спиртового раствора, содержащего вместе с меркаптанами и эфирокислоты или их соли: C2H5O—SO2ОК + СН3SK = CH3SC2H5 + K2SO4. Св-ва: Тиоэфиры суть не смешивающиеся с водой и более легкие, чем она, жидкости с отвратительным запахом. Тогда как темп. кип. обыкновенных эфиров лежит ниже т. кип. соответствующих им спиртов, Т. кипят выше отвечающих им меркаптанов (тиоспиртов, см.), напр. CH3SH кип. при +6°, а (СН3)2S — при +37°, C2H5SH кип. при 36°, а (C2H5)2S — при 92°. Точки кип. Т. идут, все повышаясь, и доходят уже у сернистого первичного нормального гептила до 298°; серн. цетил при обыкновенных условиях уже твердое воскообразное вещество с т. плавл. 57,5°. В химическом отношении Т. индифферентны, не заключая водорода, который подвергался бы замещению металлами. С галоидами и галоидными солями они дают кристаллические соединения, напр.: (C2H5)2SHgCl2, (CH3)2SHgJ2, (CnH2n+1)2SBr2. При действии крепкой азотной кислоты из Т. получаются азотнокислые сульфоокиси — (CnH2n+1)SO·HNO3, a при более энергичном и продолжительном действии сульфоны (CnH2n+1)2SO2. Дитиоэфиры CnH2n+1S2CmH2m+1 получаются при перегонке солей алкилсерной кислоты с двусернистым калием: 2С2Н5O—SO2—OK + K2S2 = (С2Н5)S2 + 2K2SO4 а также взаимодействием галоидалкилов с двусернистым калием, наконец, при окислении меркаптанов воздухом, крепкой серной кислотой, хлористым сульфурилом, при действии йода на меркаптид натрия. Смешанные ди-Т. получены действием брома на смесь двух меркаптанов: С2Н5SH + С5Н11SH + Br2 = 2ΗΒr + С2Н5—S—S—C5H11. С2Н5—SS—C2Н5 + 2Н = 2С2Н5SH. При окислении слабой азотной кислоты ди-Т. дают эфиры тиосульфоновой кислоты. 2С2Н5SH + S2Cl2 = 2HCl + С2Н5S4С2Н5. Сопоставление св-в. 19.Сульфокислоты алифатического ряда. Способы их получения.Сульфоокисление алканов и циклоалканов:таким способом получают алкано- и циклоалкансульфоновые к-ты: R-H→R-SO3H Гидролиз сульфонилхлоридов: CH3CH2CH3 →CH3-CH-CH3 +HCl │SO3H пропан 2-пропансульфонилхлорид 34. Перегруппировки амидов (Гофман) ГОФМАНА РЕАКЦИИ. 1) Превращение амидов карбоновых к-т в первичные амины с элиминированием СО2, происходящее под действием гипогалогенитов щелочных металлов (перегруппировка Гофмана, расщепление амидов по Гофману): К водному щелочному р-ру гипогалогенита прибавляют амид: послед. нагревание до 40-80°С завершает р-цию. Для увеличения выхода аминов из амидов высших жирных к-т р-цию проводят в спиртовом р-ре. Образующиеся при этом уретаны легко гидролизуются в амины. Первая стадия Гофмана реакции-синтез N-галогенамида, образующего под действием щелочей нестойкую соль I: Стадия, определяющая скорость Гофмана реакции,-отщепление галогена с образованием нитрена II, к-рый стабилизируется перегруппировкой в изоцианат: Последний при взаимод. с Н2О через карбаминовую к-ту превращ. в амин: Установлено, что мигрирующая группа сохраняет конфигурацию. Нестойкий анион амида III может взаимод. с соед., содержащими электроф. кратные связи, напр.: 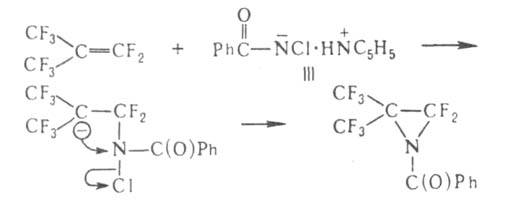 Амиды  С помощью Гофмана реакции можно получить с хорошими выходами алифатич., жирно-ароматич., ароматич. и гетероциклич. амины,диамины и аминокислоты; в пром-сти Гофмана реакция используется для синтеза антраниловой к-ты. Р-ция открыта А. В. Гофманом в 1881. 2) Разложение четвертичных аммониевых оснований, имеющих в углеродной цепи атом Н в Обычно проводят путем упаривания водного или спиртового р-ра гидроксида тетраалкиламмония (часто в вакууме) с постепенным повышением т-ры до 100-150 °С. Использование смеси безводных ДМСО и ТГФ позволяет снизить т-ру р-ции до комнатной. Если атомN связан с разл. алкильными заместителями, то в осн. образуется олефин с наименьшим числом алкильных групп у двойной связи(правило Гофмана), напр.: Заместители при В случае двух сопряженных связей в Гофмана реакция, как правило, протекает по механизму бимолекулярного элиминирования и стереоспецифична, в нек-рых случаях-по внутримолекулярному циклич. механизму, напр.: 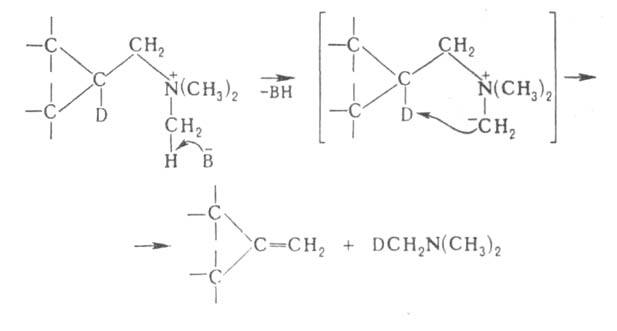 где В--основание. Основные побочные процессы при Гофмана реакции-образование спиртов, а также простых эфиров, эпоксидов, производных ТГФ ициклопропана. В р-цию, подобную Гофмана реакции, вступают соли сульфония и фосфония, напр.: 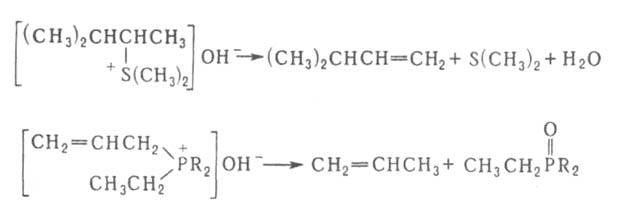 Гофмана реакцию применяют для синтеза непредельных соединений. Последовательное метилирование аминов и их расщепление (т. наз. исчерпывающее метилирование) используется для идентификации аминов. Р-ция открыта А. В. Гофманом в 1851. 20. Методы синтеза алифатических нитросоединений. Механизмы реакций. НИТРОСОЕДИНEНИЯ (С-нитросоединения), содержат в молекуле одну или неск. нитрогрупп, непосредственно связанных с атомом углерода. Известны также N- и О-нитро-соединения .Нитрогруппа имеет строение, промежуточное между двумя предельными резонансными структурами: При действии оснований на первичные и вторичные нитросоединения образуются соли нитросоединений; амбидентные анионы солей в р-циях с электрофилами способны давать как О-, так и С-производ-ные. Так, при алкилировании солей нитросоединений алкилгалогенидами, триалкилхлорсиланами или R3O+BF-4 образуются продукты О-алкилирования. Последние м.б. получены также при действии диазометана либо N,О-бис-(триметилсилил)аце-тамида на нитроалканы с рКа < 3 или нитроновые к-ты, напр.: 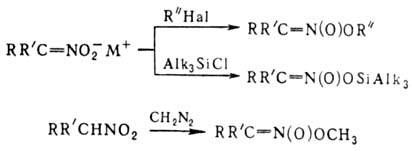 Ациклич. алкиловые эфиры нитроновых к-т термически нестабильны и распадаются по внутримол. механизму: 21. Строение нитрогруппы (семиполярная связь, мезомерия). Свойства алифатических нитросоединений. Таутомерия, отношение к щелочам и азотистой кислоте. Конденсация с карбонильньми сое динениями. Ацидолиз первичных алифатических нитросоединений , превращение их в нитрилы.Нитрогруппа - одна из наиб.сильных электроноакцепторных групп и способна эффективно делокализоватьотрицат. заряд. В ароматич. соед. в результате индукционного и особенно мезомерного эффектов она влияет на распределение электронной плотности: ядро приобретает частичный положит. заряд, к-рый локализован гл. обр. в орто- и пара-положениях; константыГаммета для группы NO2sм 0,71, sn 0,778, s+n 0,740, s-n 1,25. Т. обр., введение группы NO2 резко увеличивает реакц. способность орг. соед. по отношению к нуклеоф. реагентам и затрудняет р-ции с электроф. реагентами. Это определяет широкое применение нитросоединений в орг. синтезе: группу NO2 вводят в нужное положение молекулы орг. соед., осуществляют разл. р-ции, связанные, как правило, с изменением углеродного скелета, и затем трансформируют в др. ф-цию или удаляют. В ароматич. ряду часто используют и более короткую схему: нитрование-трансформация группы NO2.Ациклич. алкиловые эфиры нитроновых к-т термически нестабильны и распадаются по внутримол. механизму:  Кето-енольная таутомерия и, как следствие, двойственная реакц. способность (р-ции по атомам О или C) обусловливают широкие синтетич. возможности b-дикарбонильных соединений (см. Дикарбонилъные соединения). 22.Методы синтеза алифатических аминов .Алифатические амины Амины - производные аммиака, в которых атомы водорода замещены на углеводородные радикалы. Амины классифицируют по числу атомов водорода, замещенных на углеводородные радикалы. Различают первичные R-NH2, вторичные R2-NH и третичные R3-N амины. Известны и соединения с четвертичным атомом азота - соли аммония - R4N+X В этом случае азот несет положительный заряд. |