Педиатрия вич спид. детский спид вич и не только. Анатомофизиологические особенности эндокринной системы у детей. Методика обследования эндокринных желез. Семиотика синдромов гипер и гипофункций отдельных эндокринных желез и заболеваний эндокринной системы у детей.
 Скачать 264.67 Kb. Скачать 264.67 Kb.
|

Поджелудочная железаПоджелудочная железа относится к железам со смешанной функцией. Эндокринная функция осуществляется за счет продукции гормонов панкреатическими островками Лангерганса (Па́уль Лангерга́нс - Paul Langerhans; 1849-1888 -- немецкий анатом и гистолог). Островки расположены преимущественно в хвостовой части железы, и небольшое их количество находится в головном отделе. В островках имеется несколько типов клеток: альфа-клетки (α) составляют 15-20% пула островковых клеток, вырабатывают глюкагон (естественный антагонист инсулина); бета-клетки (β) составляют 65-80% пула островковых клеток, продуцируют инсулин; дельта-клетки (δ) составляют 3-10% пула островковых клеток, синтезируют соматостатин, который угнетает секрецию инсулина и глюкагона; ПП-клетки составляют 3-5% пула островковых клеток , вырабатывают панкреатический полипептид - антагонист холецистокинина, подавляет секрецию поджелудочной железы и стимулирует секрецию желудочного сока. Инсулин влияет на все виды обмена веществ, но прежде всего на углеводный. Под воздействием инсулина происходит уменьшение концентрации глюкозы в плазме крови (гипогликемия). Инсулин повышает проницаемость клеточной мембраны для глюкозы, что усиливает ее утилизацию. Инсулин также способствует превращению глюкозы в гликоген в печени и мышцах (гликогенез). Он активирует ферменты, участвующие в превращении глюкозы в гликоген печени, и ингибирует ферменты, расщепляющие гликоген. Кроме того, инсулин угнетает активность ферментов, обеспечивающих глюконеогенез, за счет чего тормозится образование глюкозы из аминокислот. Инсулин стимулирует синтез белка из аминокислот и уменьшает катаболизм белка. Инсулин регулирует жировой обмен, усиливая процессы липогенеза: способствует образованию жирных кислот из продуктов углеводного обмена, тормозит мобилизацию жира из жировой ткани и способствует отложению жира в жировых депо. Образование инсулина регулируется уровнем глюкозы в плазме крови. Гипергликемия способствует увеличению выработки инсулина, гипогликемия уменьшает образование и поступление гормона в кровь. Некоторые гормоны желудочно-кишечного тракта, такие как желудочный ингибирующий пептид, холецистокинин, секретин, увеличивают выход инсулина в кровь. Блуждающий нерв и ацетилхолин усиливают продукцию инсулина, симпатические нервы и норадреналин подавляют секрецию инсулина. Антагонистами инсулина по характеру действия на углеводный обмен являются глюкагон, АКТГ, соматотропин, глюкокортикоиды, адреналин, тироксин. Сахарный диабет (diabetesmellītus) - группа заболеваний, развивающихся вследствие абсолютной или относительной недостаточности инсулина. Термин «диабет» впервые был использован греческим врачом Деметриосом из Апамании (II век до н.э.), происходит от др.-греч. διαβαίνω, что означает «перехожу, пересекаю». В то время было представление о диабете как о состоянии, при котором человек непрерывно теряет жидкость и её восполняет, «как сифон», что относится к одному из основных симптомов диабета -- полиурии. В 1675 году Томас Уиллис показал, что при полиурии моча может быть «сладкой», а может быть и «безвкусной». В первом случае он добавил к слову диабет (лат. diabetes) слово mellitus, что с латинского означает «сладкий, как мёд» (лат. diabetes mellitus), а во втором -- «insipidus», что означает «безвкусный». Инсулинзависимый сахарный диабет (ИЗСД) - сахарный диабет I типа- болезнь, вызванная разрушением β-клеток островков поджелудочной железы. Как правило, ИЗСД поражает детей, подростков и молодых людей (отсюда его прежнее название: ювенильный диабет), но может начинаться в любом возрасте. Современное название болезни - инсулинзависимый сахарный диабет - указывает на пожизненную потребность больных в инсулине. Генетическая предрасположенность к ИЗСД обусловлена несколькими генами. Главная причина ИЗСД - разрушение β-клеток. В большинстве случаев это разрушение β-клеток имеет аутоиммунную природу. Аутоиммунную реакцию против β-клеток у лиц с генетической предрасположенностью к диабету могут индуцировать вирусные инфекции(вирус Коксаки, вирусы эпидемического паротита, краснухи, ветряной оспы, кори, цитомегаловирус), а также токсические вещества, избирательно поражающие β-клетки и индуцирующие аутоиммунную реакцию. Как правило, разрушение β-клеток происходит медленно и постепенно и сначала не сопровождается нарушениями углеводного обмена. Эту фазу развития болезни называют латентным диабетом или доклиническим периодом диабета. Когда погибает 80-95% β-клеток, возникает абсолютный дефицит инсулина, развиваются тяжелые метаболические нарушения и наступает клинический период болезни . Классические симптомы диабета: полиурия - резко увеличенный объем выделяемой мочи. Гипергликемия приводит к усиленной экскреции глюкозы. Осмотически активная глюкоза увлекает за собой воду. Для детей младшего возраста характерно недержание мочи. полидипсия - употребление большого количества воды, так как потеря воды (полиурия) вызывает постоянную жажду. полифагия - употребление большого количества пищи. Постоянное чувство голода вызвано нарушением утилизации глюкозы и потерей глюкозы с мочой. потеря веса- признак, патогномоничный для ИЗСД (у больных с инсулиннезависимым сахарным диабетом обычно наблюдается избыточный вес). Потеря веса к моменту клинического проявления диабета особенно характерна для детей. Главные причины - экскреция глюкозы (потеря калорий) и полиурия. Другие клинические признаки диабета: сухость слизистых и кожи вызвана потерей воды; диабетический «румянец» - порозовение кожи на щеках, подбородке и надбровных дугах; утомляемость и слабость обусловлены нарушением утилизации глюкозы и сдвигами электролитного обмена; частые инфекции, особенно кожные; Основные лабораторные критерии диагностики диабета - гипергликемия, глюкозурия, кетонурия. Аутоантитела к островковым клеткам, аутоантитела к инсулину присутствуют в сыворотке у 80-95% больных с впервые выявленным диабетом. К сожалению, в 20% случаев первыми проявлениями диабета у детей оказываются диабетический кетоацидоз или кетоацидотическая кома. При диабетическом кетоацидозе в крови накапливаются кетокислоты (кетоновые тела): ацетон, ацетоуксусная кислота, β-оксимасляная кислота, что приводит к ацидозу. Клинические проявления: гипотермия, гипорефлексия, снижение тургора и сухость кожи, нарушение дыхания (Куссмауля), тахикардия, тошнота и рвота, запах ацетона (или фруктовый) изо рта. При прогрессировании ацидоза возникает артериальная гипотония, сопор и кома. Неотложная помощь при диабетическом кетоацидозе (прекоматозное состояние): 1. Укрыть ребенка одеялом. 2. Промыть желудок раствором 2% соды. 2. Поставить очистительную, а затем лечебную клизму с теплым раствором 2% соды в объеме 150-200 мл. 3. Провести инфузионную терапию физиологическим раствором, подогретым до 37º С. 4. Инсулинотерапия проводится из расчета 0,1 Ед/кг/час (детям дошкольного возраста - 0,05 Ед/кг/час). Кроме классическогоинсулинзависимого сахарного диабета (диабет I-го типа), дети могут заболевать и диабетом инсулиннезависимым (II-го типа), который может быть вторичным, возникающим на фоне муковисцидоза или первичным на фоне ожирения. Надпочечники В надпочечниках различают два слоя, или вещества: корковое и мозговое, причем первое составляет примерно 2/3 общей массы надпочечника. Корковый слой имеет мезодермальное происхождение, мозговой слой развивается из зачатка симпатического ганглия. Синдром и болезнь Кушинга имеют одинаковые клинические проявления в связи с избыточной продукцией глюкокортитоидов в коре надпочечников. При первичном поражении надпочечников говорят о синдроме Кушинга. При патологии гипоталамо-гипофизарной системы с избыточной продукцией адренокортикотропного гормона говорят о болезни Кушинга. Американский хирург Харви Кушинг в 1932 году описал клинический синдром, который назвал «гипофизарный базофилизм». Клинические проявления: задержка роста, склонность к геморрагическим высыпаниям (петехии, экхимозы). На фоне повышенной секреции глюкокортикоидов отмечается остеопороз, ожирение с отложением жира над ключицами, на шее, затылке, лице, которое становится круглым (лунообразное лицо), стрии на животе, груди и внутренней поверхности бедер.  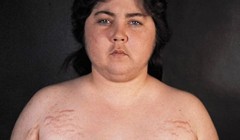 Следствием избыточного синтеза альдостерона является артериальная гипертония, общая мышечная слабость. Избыточное производство андрогенов вызывает появление угрей, гирсутизма (лат. hirsutus - волосатый) - избыточное оволосение по мужскому типу. Нередко отмечается вирилизация (лат. virilis мужской, свойственный мужчине; син.: андрогенизация, маскулинизация) - процесс проявления мужских черт (тип телосложения, оволоcение, тембр голоса и др.) у женщин под влиянием гормонов. При дополнительном обследовании определяется повышение уровня глюкокортикоидов в крови и нарушение регуляции в гипоталамо-гипофизарно-надпочечниковой системе. Недостаточность надпочечников может развиться при поражении надпочечников (первичная) или при снижении секреции адренокортикотропного гормона (АКТГ) гипофиза (вторичная). Надпочечниковая недостаточность бывает острой (ОНН) - синдром Уотерхауза--Фридериксенаи хронической (ХНН) - аддисонова болезнь. Синдром Уотерхауза-Фридериксена (R. Waterhouse, англ. врач; С. Friderichsen, дат. педиатр) наблюдается при врожденной гипоплазии надпочечников; врожденном адреногенитальном синдроме; различного происхождения кровоизлияниях в надпочечники; при заболеваниях, для которых характерны кровоизлияния и некроз надпочечников (менингококцемия, сепсис, дифтерия). Наиболее часто синдром Уотерхауза--Фридериксена наблюдается после кровоизлияния в надпочечники при менингококцемии - генерализованной менингококковой инфекции. Отмечается озноб и гипертермия, на коже появляются многочисленные петехии и кровоподтеки, кожа местами приобретает окраску, напоминающую трупные пятна. Артериальное давление прогрессивно падает. Пульс частый, нитевидный. Появляются рвота, судороги, одышка. Больные впадают в быстро прогрессирующую прострацию с потерей сознания. Внезапная смерть является следствием остро развившейся недостаточности надпочечников. Причинами Аддисоновой болезни (по имени описавшего её впервые в 1855 английского врача Т. Аддисона) в настоящее время наиболее часто являются аутоиммунный процесс и генетические факторы, реже хронические интоксикации, токсоплазмоз, кистозное перерождение надпочечников. Вторичная надпочечниковая недостаточность возникает при поражении головного мозга с последующим снижением продукции АКТГ. Характерны астенизация, утомляемость, снижение работоспособности и успеваемости, похудание, гипогликемия, мышечная атрофия, анорексия, тошнота, тяга к соленой пище, жажда, периодические рвота и понос. Характерны типичные изменения кожи и слизистых оболочек, кожа имеет различные оттенки - от светло-коричневого до темно-бронзового, иногда сероватого или желтоватого. Аддисонова болезнь подтверждается низким уровнем гидрокортизона (кортизола) в сыворотке крови, изменением его суточного ритма, когда отсутствует характерный подъем уровня в ранние утренние часы. Врожденная гиперплазия коры надпочечниковчаще всего обусловлена недостаточностью 21-гидроксилазы. Известны две классические формы болезни: простая вирилизующая и сольтеряющая. Главная причина болезни - нарушение синтеза кортизола. Постоянный дефицит кортизола по принципу отрицательной обратной связи стимулирует секрецию АКТГ в гипофизе, что и служит причиной гиперплазии коры надпочечников. Избыток АКТГ приводит к усилению стероидогенеза. Однако кортизола по-прежнему вырабатывается недостаточно. Но параллельно усиливается выработка андрогенов. Избыток андрогенов ведет к усилению роста тела, вилиризации, нарушению формирования наружных половых органов. Отсюда другое название заболевания - адреногенитальный синдром. Другие симптомы определяются тем, повышена или снижена продукция альдостерона, что сопровождается соответственно артериальной гипертензией либо потерей организмом соли. Более 90% случаев врожденной гиперплазии надпочечников обусловлено двумя типами недостаточности 21-гидроксилазы: частичной (простая вирилизация) или полной (потеря соли организмом). Остальные случаи связаны с недостаточностью 11-β-гидроксилазы. У девочек при врожденной гиперплазии коры надпочечников на фоне повышенного уровня андрогенов в период внутриутробного развития к моменту рождения формируется женский псевдогермафродитизм.  Гермафродитизм - наличие в одном организме признаков обоих полов. Гермафродитизм бывает истинным и ложным. При истинном гермафродитизме в одном организме присутствуют первичные признаки обоих полов - половые железы мужские (яички) и женские (яичники). При ложном гермафродитизме первичные половые признаки (половые железы) сформированы по одному полу, а вторичные половые признаки - по другому. При адреногенитальном синдроме у девочки имеются яичники (первичные половые признаки), а вторичные половые признаки сформированы по мужскому типу: увеличение клитора (до формы пениса), слияние и гипертрофия половых губ (до размеров мошонки). После рождения маскулинизация прогрессирует. Преждевременно растут волосы на лобке, в подмышечных впадинах, грубеет голос, больные девочки выше своих сверстниц, костный возраст опережает хронологический, у них хорошо развита мускулатура. Если не проводится соответствующее лечение, грудные железы не развиваются и менструаций нет. Происходит ложное (не вырабатываются половые клетки в половых железах) преждевременное половое созревание по гетеросексуальному (мужскому) типу. При сольтеряющей форме вирилизация выражена в большей степени, чем при варианте без потери соли. У мальчиков при сольтеряющей форме адреногенитального синдрома появляются рвота, шок и электролитный дисбаланс в возрасте 7-10 дней. У мальчиков при простой форме нарушение проявляется признаками ложного преждевременного полового созревания по изосексуальному (мужскому) типу. Данное раннее половое созревание является ложным, так как не формируется сперматогенез. Повышенный уровень андрогенов по принципу обратной связи приводит к торможению выработки гонадотропных гормонов в гипофизе, что является непосредственной причиной отсутствия сперматогенеза. При рождении ребенок выглядит нормальным, но признаки преждевременного полового и соматического развития могут проявиться уже на первом полугодии жизни или развиваются более медленно и становятся очевидными только в возрасте 4-5 лет и позднее. К числу таких признаков относятся: увеличение полового члена, мошонки, появление волос на лобке, угри, запах пота, понижение тембра голоса. Яички нормального размера, но по сравнению с увеличенным половым членом кажутся маленькими. Мышечная система хорошо развита, костный возраст опережает хронологический. Умственное развитие не страдает, но из-за особенностей физического развития возможны аномалии поведения. Преждевременное закрытие эпифизов приводит к раннему закрытию зон роста и в итоге к низкорослости. Нарушения пола и полового развития Нарушения пола и полового развития у детей очень многообразны и встречаются часто, особенно у мальчиков. Преобладающими являются расстройства сроков полового созревания, чаще его отставание, реже опережение. Нормальное развитие ребенка и полноценная его социально-психологическая адаптация возможны только в том случае, если имеется полное совпадение пола генетического, гонадного, соматического и психологического. Это состояние называютизосексуальностью. При аномальном формировании пола этого единства уже не будет, и для определения этого состояния используют термины«гетеросексуальность» или«интерсексуальность». Врачебный контроль за течением процесса полового созревания, прежде всего, включает в себя оценку возраста начала появления пубертатных сдвигов. Наиболее ранней границей появления признаков полового созревания можно считать для девочек возраст около 8-8,5 лет, для мальчиков - 10-10,5 лет. Если наблюдается более раннее начало, ребенок подлежит специальному эндокринологическому обследованию. Преждевременное половое развитие - это появление некоторых или всех вторичных половых признаков (а в некоторых случаях - и наступление половой зрелости) у девочек младше 8 лет или у мальчиков младше 9 лет. При формирование вторичных половых признаков изосексуальное (соответствующее генетическому и гонадному полу ребенка) и завершенное - обусловлено активацией гипоталамуса и гиперсекрецией гонадотропных гормонов. При истинном преждевременном половом развитии у девочек последовательность событий обычно такая же, как в норме: сначала наступает телархе (увеличение молочных желез), затем адренархе (оволосение андрогензависимых зон), затем ускорение роста и, наконец, менархе (менархе - греч. men, месяц и arche, начало - первая менструация). Однако у некоторых больных телархе и менархе могут наступить задолго до адренархе. Это объясняется тем, что секреция эстрогенов в яичниках и секреция андрогенов в надпочечниках регулируются независимо. Половое развитие у мальчиков преждевременное истинное обусловлено гиперфункцией центрального звена гипоталамо-гипофизарно-гонадной системы. Причины: ранняя активация импульсной секреции гонадолиберина в гипоталамусе, ранняя секреция гонадотропных гормонов в гипофизе, другие нарушения регуляции в гипоталамо-гипофизарной системе. Истинное преждевременное половое развитие всегда полное (т. е. включает как вирилизацию, так и стимуляцию сперматогенеза). Ложное преждевременное половое развитие вызвано другими причинами, обычно остается незавершенным (нет сперматогенеза у мальчиков и овуляции у девочек) и может быть как изо-, так и гетеросексуальным. Половое развитие преждевременное ложное не зависит от секреции гонадолиберина и не связано с первичными нарушениями секреции гонадотропных гормонов. Половое развитие у девочек преждевременное ложное гетеросексуальное Эта форма преждевременного полового развития характеризуется появлением мужских вторичных половых признаков у девочек из-за избытка андрогенов . Самая распространенная причина - простая вирилизирующая форма адреногенитального синдрома. Ложное преждевременное половое развитие у мальчиков обусловлено автономной гиперсекрецией андрогенов или гиперсекрецией хорионического гонадотропина. В отличие от истинного преждевременного полового развития , ложное преждевременное половое развитие неполное, т. е. не сопровождается стимуляцией сперматогенеза. Самая распространенная причина ложного преждевременного полового развития мальчиков - простая вирилизирующая форма адреногенитального синдрома. Задержка полового развития Изолированный дефицит гонадотропных гормонов в большинстве случаев обусловлен врожденным нарушением секреции гонадолиберина, реже - врожденной или приобретенной недостаточностью гонадотропных клеток аденогипофиза. В любом случае врожденный изолированный дефицит гонадотропных гормонов приводит к задержке полового развития (оно не начинается или не завершается) и проявляется симптомами вторичного гипогонадизма. Задержка полового развития также может быть на фоне задержки физического развития, белково-калорийной недостаточности и гиповитаминозов, хронических соматических заболеваний, чрезмерных физических нагрузок. Конституциональная задержка полового развития - это вариант нормы. Она обусловлена запаздыванием активации гипоталамо-гипофизарно-гонадной системы . Как и при изолированном дефиците гонадотропных гормонов, уровни ЛГ, ФСГ и тестостерона снижены. Из-за этого половое развитие начинается в 15 лет и позже. |