Патфиз ч.3. Патфиз ч. Частнаяпатология
 Скачать 13.12 Mb. Скачать 13.12 Mb.
|

|
СИМПТОМАТИЧЕСКОЕ ЛЕЧЕНИЕ Симптоматическая терапия имеет целью облегчение состояния пациента. Для этого устраняют неприятные, тягостные ощущения (болевые, психоэмоциональные и др.), а также проводят мероприятия по устранению недостаточности функции органов и физиологических систем. Профилактика Профилактика повторного развития ДВС — ликвидации или предупреждении возникновения условий, провоцирующих развитие ДВС (терапия основного заболевания, введение гепарина при гиперкоагуляции, повторные трансфузии свежезамороженной плазмы). ПРОГНОЗ Прогноз во многом зависит от эффективности терапии основного заболевания, своевременности диагностики ДВС-синдрома, адекватности лечебных мероприятий. Летальность при ДВС составляет 40–60%. Основные причины смерти: острая почечная недостаточность, дыхательная недостаточность, кровоизлияние в мозг, надпочечники, острая кровопотеря, приводящая к развитию шока и комы. ГЕМОБЛАСТОЗЫ Гемобластозы — опухоли, возникающие из кроветворных клеток гемопоэтической ткани. Гемобластозы занимают первое место как причина смерти среди всех болезней системы крови. Гемобластозы подразделяют на лейкозы (опухоли, диффузно — системно –поражающие гемопоэтические клетки костного мозга), лимфомы (внекостномозговые плотные, растущие в виде узла или нескольких узлов, опухоли из лимфопролиферативных кроветворных клеток) и миелопролиферативные новообразования (миеломы). В клинической практике используют не родовое понятие «лейкоз», а названия конкретных нозологических форм лейкоза (разных его стадий и определённых иммуно-, фено- и генотипов), каждая из которых подразумевает конкретную программу лечения. Так, согласно МКБ–10, к злокачественным новообразованиям «лимфоидной, кроветворной и родственных им тканей» (коды МКБ — C81–C96) относятся болезнь Ходжкена (лимфогранулематоз), неходжкенские лимфомы (лимфосаркомы), злокачественные иммунопролиферативные болезни (в том числе макроглобулинемия Вальденстрёма), множественная миелома, лимфоидный лейкоз (лимфолейкоз), миелоидный лейкоз (миелолейкоз), истинная полицитемия и ряд других опухолей. Для лимфоидных гемобластозов предложена классификация REAL (Revised Europian–American classification of Lymphoid neoplasms, см. статью «Классификация REAL» в приложении «Справочник терминов» на компакт диске). В клинической литературе лимфомы (растущих вне костного мозга очаговые опухоли из кроветворных клеток) обозначают как гематосаркомы (син.: лимфосаркомы). К гематосаркомам относят также миело-, эритро-, мегакариосаркомы, а также морфологически недифференцируемые гематосаркомы. При метастазировании гематосарком в костный мозг опухолевый процесс приобретает генерализованный характер. Этот феномен обозначают как лейкемизация гематосарком. Характеристики отдельных лимфом приведены в статье «Лимфомы» приложения «Справочник терминов» на компакт диске. В этом же приложении рассмотрены этиология, патогенез, проявления и принципы терапии других гемобластозов: истинной полицитемии, лимфогранулематоза (болезни Ходжкена), миеломной болезни, макроглобулинемии Вальденстрёма. ОБЩАЯ ХАРАКТЕРИСТИКА ЛЕЙКОЗОВ Лейкоз — системное опухолевое поражение гемопоэтических клеток костномозговой ткани. Для обозначения лейкозов не рекомендуется применять старый, предложенный еще Р. Вирховым, термин «лейкемия» (белокровие) в связи с тем, что к лейкозам относятся, помимо опухолей из лимфо- и миелопоэтических клеток, также и новообразования из клеток эритро- и мегакариоцитарных ростков. ЭТИОЛОГИЯ ПРИЧИНЫ Причинами лейкозов являются те же группы факторов, которые вызывают опухоли (см. главу 17 «Опухоли»). ФАКТОРЫ РИСКА • Наследственное предрасположение. Описаны доминантное и рецессивное наследование хронического лимфолейкоза (ХЛЛ), а также низкая заболеваемость этим лейкозом в одних этнических группах и высокая в других. Чаще в этих случаях наследуется не сам лейкоз, а нестабильность генома — сниженная резистентность хромосом к действию мутагенов, предрасполагающая родоначальные миелоидные или лимфоидные клетки к опухолевой (лейкозной) трансформации. • Применение лекарственных средств с цитостатическим действием. Вероятность возникновения острых лейкозов у больных, лечившихся цитостатиками, повышается в сотни раз. • Воздействие на организм проникающей радиации и/или рентгеновского излучения (в том числе- с лечебной целью). Минимальный интервал времени до возникновения лейкоза после облучения составляет 5–10 лет, а после химиотерапии — 2 года (максимум в течение 6–10 лет). ПАТОГЕНЕЗ Трансформация нормальных гемопоэтических клеток в опухолевые является результатом изменений в генетической прграмме. Основную роль при этом играют хромосомные нарушения. В большинстве случаев они определяют прогноз болезни и тип специфического лечения. Нестабильность генома лейкозных клеток приводит к появлению в первоначальном опухолевом клоне новых субклонов, среди которых в процессе жизнедеятельности организма, а также под воздействием лечения "отбираются" наиболее автономные (этот феномен получил название «опухолевая прогрессия»). Этим феноменом объясняют прогредиентность течения лейкозов, их «уход» из-под контроля цитостатиков и генерализация процесса. Этиология, патогенез, проявления и принципы лечения различных лейкозов (лейкоз острый, миелолейкоз хронический, лимфолейкоз хронический) рассмотрены в разделе «Лейкозы» настоящей главы. Характеристика отдельных видов лимфом приведена в статье «Лимфомы» приложения «Справочник терминов» на компакт диске. В этом же приложении рассмотрены этиология, патогенез, проявления и принципы терапии других гемобластозов: истинной полицитемии, лимфогранулематоза (болезни Ходжкена), миеломной болезни, макроглобулинемии Вальденстрёма. ОПУХОЛЕВЫЙ АТИПИЗМ Атипизм гемобластозов — совокупность существенных качественных и количественных отличий биологических свойств гемобластозных клеток от нормальных и других патологически изменённых (но не трансформированных) клеток и тканей. В основе формирования атипизма гемобластозов лежит процесс опухолевой прогрессии. ОПУХОЛЕВАЯ ПРОГРЕССИЯ Опухолевая прогрессия по своему существу является механизмом нарастания степени злокачественности клеток гемобластозов в результате изменений их генетической программы. Повышенная и постоянная изменчивость различных свойств гемобластозов, обусловливая их гетерогенность и автономность, создаёт условия для всё большей приспособленности их клеток к условиям среды, включая и возможность «ускользания» от цитостатической терапии. ПРИЗНАКИ ОПУХОЛЕВОЙ ПРОГРЕССИИ ГЕМОБЛАСТОЗОВ Проявления опухолевой прогрессии гемобластозов приведены на рис. 21–34. 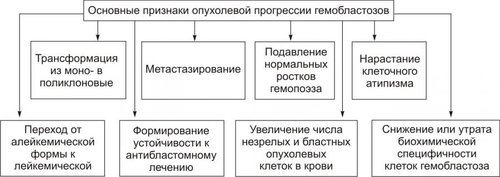 Рис. 21–34. Признаки опухолевой прогрессии гемобластозов. Процесс опухолевой прогрессии ведет к формированию и/или нарастанию степени атипизма роста, обмена, структуры и функции лейкозов. АТИПИЗМ РОСТА Проявления атипизма роста лейкозов представлены на рис. 21–35. 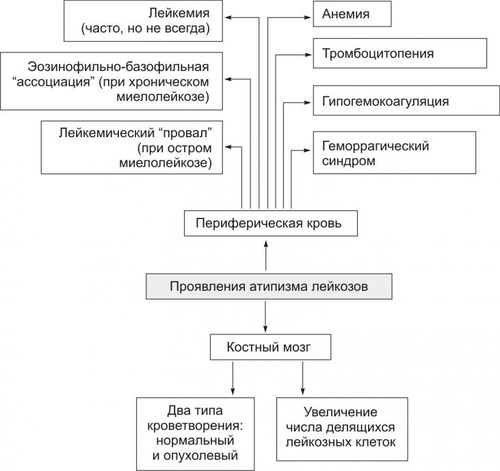 Рис. 21–35. Основные проявления атипизма роста лейкозов. • Костный мозг. † Наличие клеток, относящихся к двум качественно разным типам гемопоэза: нормальному и опухолевому. † Увеличение числа делящихся гемопоэтических клеток («омоложение» состава гемопоэтических клеток). Это сопровождается нарастанием количества атипичных бластных и молодых нормальных клеток гемопоэтической ткани. • Периферическая кровь. † Лейкемия (белокровие, от гр. leukos белый, haima кровь). Наблюдается часто, но не всегда и характеризуется увеличением количества лейкозных клеток различной степени зрелости (бластных, созревающих). В зависимости от общего количества лейкоцитов и наличия бластных клеток в единице объёма крови при лейкозах выделяют четыре формы лейкоза. ‡ Лейкемическая: число лейкоцитов превышает 30–50 ‡ Сублейкемическая: Количество лейкоцитов выше нормы (но до 30–50 ‡ Лейкопеническая: число лейкоцитов ниже нормы, сравнительно небольшое количество бластных лейкозных клеток. ‡ Алейкемическая: количество лейкоцитов в диапазоне нормы, бластные клетки отсутствуют. Атипичные лейкоциты, их бластные и молодые формы находят лишь в ткани костного мозга. † Лейкемический «провал» (лейкемические «ворота» [hiatus leukaemicus], лейкемическое «зияние»). Выявляется при остром миелобластном лейкозе и характеризуется наличием в периферической крови бластных, молодых и зрелых форм лейкозных клеток и отсутствием одной или нескольких переходных форм гемопоэза. С этим и связано название признака — лейкемический «провал». † Анемия. Является спутником большинства лейкозов, в особенности острых. † Тромбоцитопения и снижение свёртываемости крови. † Геморрагический синдром. Характеризуется частыми кровотечениями, в том числе в полости тела и полые органы (желудок, кишечник, пищевод, мочевой пузырь и др.), а также кровоизлияниями. АТИПИЗМ ОБМЕНА Атипизм обмена характеризуется отклонением от нормы биохимических характеристик клеток и/или продуктов их метаболизма. Помимо общих признаков опухолевого атипизма обмена углеводов, белков, липидов, ионов, минералов, КЩР (см. главу 17 «Опухолевый рост»), для лейкозов характерен ряд специфичных признаков. • Прекращение синтеза лейкозными клетками отдельных ферментов (например, кислой фосфатазы, миелопероксидазы) и, как следствие — катализируемых или процессов. • Пара и диспротеинемии. Парапротеинемия наблюдается при миеломной болезни, макроглобулинемии Вальденстрёма, болезни тяжёлых цепей Франклина. АТИПИЗМ СТРУКТУРЫ Характеризуется развитием признаков клеточного (см. главу 17 «Опухолевый рост») и тканевого атипизма (проявляющегося наличием двух типов клеток в гемопоэтической ткани и периферической крови — нормальных и опухолевых). Подробнее характеристику атипизма структуры лейкозов см. в учебниках по патологической анатомии. АТИПИЗМ ФУНКЦИЙ При лейкозах нарушаются функции как трансформированных (лейкозных), так и нормальных лейкоцитов. Это приводит к существенным нарушениям фагоцитарной активности, механизмов реализации клеточного и гуморального иммунитета. В совокупности указанные нарушения обусловливают развитие иммунодефицита и в том числе снижение противоинфекционной устойчивости и антибластомной резистентности организма. ХАРАКТЕРИСТИКА ОТДЕЛЬНЫХ, НАИБОЛЕЕ ЧАСТО ВСТРЕЧАЮЩИХСЯ ЛЕЙКОЗОВ. Ниже рассмотрены острые и хронические формы лейкозов как пример современного подхода к патогенезу, типированию конкретных стадий, проявлениям и принципам лечения онкогематологических заболеваний. ОСТРЫЙ ЛЕЙКОЗ Клеточным субстратом острых лейкозов являются бластные опухолевые клетки. Острый лейкоз без лечения приводит к смертельному исходу в течение нескольких недель или месяцев. При правильном и своевременном лечении прогноз для детей часто благоприятен. Острые лейкозы подразделяют на миелоидные (острый миелолейкоз), лимфоидные (острый лимфолейкоз ), монобластные (миеломонобластные), эритромиелобластные и мегакариобластные. ХРОНИЧЕСКИЙ ЛЕЙКОЗ Морфологический субстрат хронических лейкозов — относительно дифференцированные клетки кроветворной ткани. Больные могут жить без лечения в течение нескольких месяцев и лет. Хронические лейкозы нередко трансформируются в острые формы. Хронические лейкозы подразделяют на миелоидные (хронический миелолейкоз — ХМЛ), лимфоидные (хронический лимфолейкоз — ХЛЛ и волосатоклеточный лейкоз), моноцитарные (миеломоноцитарные), эритроцитарные (истинная полицитемия) и мегакариоцитарные. ОСТРЫЕ ЛЕЙКОЗЫ Острые лейкозы — злокачественные заболевания кроветворной системы, морфологический субстрат — бластные клетки. Наиболее часто диагносируются острый лимфобластный лейкоз (ОЛЛ) и острый миелоидный лейкоз. Частота Частота острых лейкозов 13,2:100 000 среди мужчин и 7,7:100 000 среди женщин. ОЛЛ чаще развивается в детском возрасте и после 40 лет. Частота острого миелоидного лейкоза одинакова во всех возрастных группах. Патогенез Патогенез острого лейкоза обусловлен пролиферацией клона опухолевых клеток с характерными цитогенетическими нарушениями, угнетением нормального кроветворения, выходом бластных клеток в кровь, метастазированием их в другие кроветворные (селезёнка, печень, лимфоузлы) и некроветворные (кожа, ЦНС, яички, лёгкие) органы. ВИДЫ ОСТРОГО ЛЕЙКОЗА Существенный момент диагностики и последующей терапии любого лейкоза — типирование заболевания у конкретного больного. Критерии типирования лейкозов представлены на рис. 21–36. 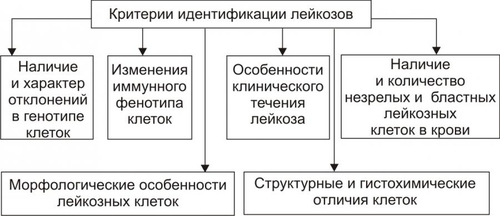 Рис. 21–36. Критерии типирования лейкозов. В основу классификаций острых лейкозов положены внешний вид и цитохимические особенности бластных клеток, их иммунофенотип и генетические особенности. Так, франко-американо-британская (FAB) классификация основана на оценке морфологии лейкозных клеток (строение ядра, соотношение размеров ядра и цитоплазмы). Острый миелоидный лейкоз Виды острого миелоидного лейкоза приведены в табл. 21–6. Таблица 21-6. Варианты острых миелоидных лейкозов (классификация ВОЗ, 1999).
ОМЛ — острый миелоидный лейкоз Об уровне дифференцировки лимфоидных клеток свидетельствует экспрессия на поверхности их ядерной мембраны, в цитоплазме и на цитоплазматической мембране различных Аг, «кластеров дифференцировки», обозначаемых аббревиатурой CD (от англ. cluster of differentiation). Иммунофенотипическая характеристика миелобластных лейкозов приведена в таблице 21-7. Таблица 21–7. Иммунофенотипическая характеристика миелобластных лейкозов.
По: «Внутренние болезни», М: ГЭОТАР–МЕД, 2001. Условные обозначения: «+» — выраженная экспрессия Аг; «+/–» — вариабельная экспрессия Аг; «–» — отсутствие экспрессии Аг Острый лимфоидный лейкоз Развитие B- и T-лимфоцитов и типы ОЛЛ рассмотрены на рис. 21–37, а иммунологические и цитогенетические варианты ОЛЛ приведены в таблице 21–8. 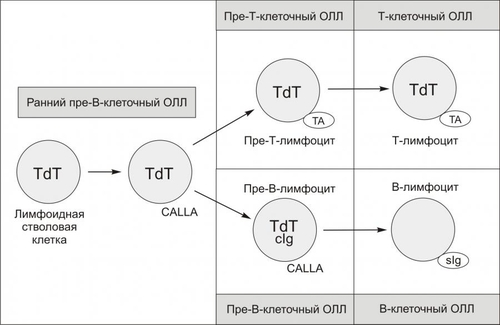 Рис. 21–37. Развитие B- и T-лимфоцитов и типы острого лимфобластного лейкоза. ОЛЛ — острый лимфобластный лейкоз, CALLA — общий Аг ОЛЛ; TdT — терминальная дезоксинуклеотидилтрансфераза; ТА — Аг тимуса; cIg — цитоплазматический Ig; sIg — мембранный Ig Наиболее значимыми CD-маркёрами для определения типа ОЛЛ являются: • В-клеточные Аг CD79a, CD79b, CD19, CD20, CD22, цитоплазматические и поверхностные IgM; • Т-клеточные Аг CD2, CD3, CD4, CD8, CD5, CD7; • Аг стволовых клеток CD34 и TdT (терминальная дезоксинуклеотидилтрансфераза); • Аг СD10 (CALLA, от англ. common acute lymphoid leukemia antigen, нейтральная эндопептидаза), свойственный определённой стадии дифференцировки предшественников В-клеток. Таблица 21–8. Иммунологические и цитогенетические варианты острых лимфобластных лейкозов.
Условные обозначения: cIg — цитоплазматический Ig; sIg — мембранный Ig; TdT —терминальная дезоксинуклеотидилтрансфераза; «+» — выраженная экспрессия Аг; «+/–» — вариабельная экспрессия Аг; «–» — отсутствие экспрессии Аг ПРОЯВЛЕНИЯ Начало заболевания, как правило, внезапное. Тяжёлое состояние больного может быть обусловлено выраженной интоксикацией, геморрагическим синдромом (результат тромбоцитопении), дыхательной недостаточностью (вследствие сдавления дыхательных путей увеличенными внутригрудными лимфатическими узлами). Возможно и постепенное развитие заболевания. • Анемический (гипоксический) синдром: бледность, одышка, сердцебиение, сонливость. • Снижение резистентности к инфекциям (бактериальным, грибковым и вирусным). У пациентов с лейкозами выявляют как лёгкие (локальные) формы инфекций (например, кандидозные стоматиты, гингивиты, поражения слизистых оболочек, вызванные вирусом простого герпеса), так и тяжёлые генерализованные процессы (пневмонии, сепсис). • Геморрагический синдром. При осмотре пациентов обнаруживают петехии и экхимозы на коже (самопроизвольные, в местах инъекций или механического трения). Возможны тяжёлые носовые и внутренние кровотечения (метроррагии, желудочно-кишечные кровотечения, кровоизлияния в мозг). • ДВС-синдром. • Болезненность костей. В наибольшей мере выражена в трубчатых костях и позвоночнике, в области суставов (артралгии). Обусловлена опухолевой гиперплазией костномозговой гемопоэтической ткани. • Лимфаденопатия. Возможно увеличение любой группы лимфатических узлов в связи с пролиферацией в них лейкозных лимфоидных клеток. • Печень и селезёнка увеличены. Увеличение их размеров связано с метастазированием лейкозных клеток в эти органы и образованием экстрамедуллярных очагов гемопоэза в них. • Нейролейкемия. Поражение ЦНС (нейролейкемия) возникает особенно часто при ОЛЛ и значительно ухудшает прогноз. Возникновение нейролейкемии обусловлено метастазированием лейкозных клеток в оболочки головного и спинного мозга или в вещество мозга. • Гипертрофия тимуса обусловлена метастазами лейкоза в вилочковую железу. Может вызвать сдавление органов средостения. ЛАБОРАТОРНАЯ И ИНСТРУМЕНТАЛЬНАЯ ДИАГНОСТИКА Анализ крови Подозрение на лейкоз должно возникать при наличии клинических симптомов и изменений в периферической крови: нормохромная нормоцитарная анемия; количество лейкоцитов может быть различным — низким (менее 5 Пункция костного мозга Пункция костного мозга — основной метод исследования при лейкозах. Его применяют с целью подтверждения диагноза и идентификации (морфологической, иммунофенотипической, цитогенетической) типа лейкоза. Миелограмма (количественная характеристика всех клеточных форм костного мозга) при острых лейкозах характеризуется увеличением содержания бластных клеток более 5% и до тотального бластоза. Структура бластов при этом различна в зависимости от типа лейкоза. Выявляется увеличение переходных форм клеток, лимфоцитоз. Красный росток кроветворения угнетён (за исключением острого эритромиелоза). Мегакариоциты отсутствуют или количество их незначительно (за исключением острого мегакариобластного лейкоза). Цитохимическое исследование — основной метод диагностики форм острых лейкозов. Его проводят с целью выявления специфических для различных бластов ферментов. Так, при ОЛЛ определяется положительная ШИК-реакция на гликоген, отрицательная реакция на липиды, пероксидазу, хлорацетат эстеразу. При острых миелобластных лейкозах — положительная реакция на миелопероксидазу, липиды, хлорацетат эстеразу. Иммунофенотипирование бластов позволяет определить с помощью моноклональных АТ наличие или отсутствие кластеров дифференцировки бластных клеток (CD-маркёры). Его проведение в первую очередь необходимо для точной диагностики ОЛЛ (см. табл. 21–7 и рис. 21–36), а также в случаях невозможности дифференциальной диагностики острых морфологически недифференцируемых лимфобластных и миелобластных лейкозов. Цитогенетическое исследование лейкозных клеток позволяет определить хромосомные аномалии, уточнить диагноз и прогноз. ЛЕЧЕНИЕ ОСТРЫХ ЛЕЙКОЗОВ • Специфическая химиотерапия. Направлена на достижение и закрепление ремиссии заболевания. Состоит из нескольких этапов. Различна для лимфобластного и миелобластного лейкозов и проводится по стандартным схемам (в зависимости от результатов типирования заболевания). • Сопутствующая (сопроводительная) терапия. Её проводят для борьбы с инфекциями, обусловленными агранулоцитозом, для снижения интоксикации при лизисе опухолевых лейкоцитов, для уменьшения побочных токсических эффектов химиотерапевтических препаратов. • Заместительная терапия. Необходима при угрожающей тромбоцитопении, тяжёлой анемии, нарушениях свертывания крови. • Трансплантация стволовых кроветворных клеток или костного мозга. Общие принципы сопутствующего лечения Профилактика инфекций — главное условие выживания пациентов с нейтропенией, возникшей вследствие химиотерапии. • Необходима полная изоляция пациента при количестве лейкоцитов менее 1 • При повышении температуры тела проводят клиническое и бактериологическое исследования и немедленно начинают лечение комбинациями антибиотиков широкого спектра действия: цефалоспоринами, аминогликозидами, полусинтетическими пенициллинами, меропенемом (меронем), имипенемом+циластатином (тиенам). При вторичных подъёмах температуры тела, возникших после лечения антибиотиками, обычно эмпирически назначают противогрибковые средства (флуконазол, амфотерицин В). При пневмоцистных пневмониях применяют ко-тримоксазол (бисептол) в дозе 240 мг/кг в/в, при цитомегаловирусных инфекциях — ганцикловир. • Для профилактики и лечения нейтропении можно назначить препараты гранулоцитарного [филграстим (нейпоген)] и гранулоцитарно-макрофагального [ленограстим (граноцит)] колониестимулирующих факторов. При острых миелобластных лейкозах эти препараты противопоказаны. Методы заместительной терапии • Трансфузия эритроцитарной массы. Показания: содержание Hb менее 70 г/л при клинических проявлениях анемии (одышка, сердцебиение в покое). • Трансфузия тромбоцитарной массы или тромбоконцентрата. Показания: кровоточивость на фоне содержания тромбоцитов менее 20 • Трансфузии компонентов крови совершаются всегда по жизненным показаниям. • Трансплантация стволовых клеток крови или костного мозга. Трансплантация стволовых клеток крови или костного мозга — аллогенная трансплантация — метод выбора при прогностически неблагоприятных острых лейкозах в первой ремиссии, во второй и последующих ремиссиях острых лейкозов и при неполных ремиссиях (бластоз в костном мозге не более 20%) — аллогенная трансплантация. Оптимальный донор — однояйцовый близнец или сибс. Чаще используют доноров с 35% совпадением по Аг HLA. При отсутствии совместимых доноров используют аутотрансплантацию костного мозга, взятого в период ремиссии. В настоящее время трансплантация стволовых клеток применяется чаще трансплантации костного мозга. Главное осложнение — реакция «трансплантат против хозяина». Развивается вследствие пересадки Тлимфоцитов донора, распознающих Аг реципиента как чужеродные и вызывающих иммунную реакцию против них. Острая реакция развивается в течение 7–100 дней после трансплантации, отсроченная — через 6–12 мес. Основные органы-мишени — кожа (дерматит), ЖКТ (диарея) и печень (токсический гепатит). Для профилактики и лечения реакции «трансплантат против хозяина» применяют селективный иммунодепрессант — циклоспорин в дозе 3 мг/кг/сут до купирования клинических проявлений с постепенной последующей отменой препарата (не более 25% дозы препарата в неделю). На течение посттрансплантационного периода влияют также подготовительные схемы лечения, развитие интерстициальной пневмонии, контаминированность конкретного больного вирусами гепатитов, цитомегаловирусом, вирусом Эпстайна–Барр. ПРОГНОЗ Острый лимфобластный лейкоз У детей в 95% и более случаев наступает полная ремиссия. У 70–80% больных детей болезнь не проявляется в течение 5 лет, их считают здоровыми. При возникновении рецидива в большинстве случаев можно достичь второй полной ремиссии. Больные со второй ремиссией — кандидаты на трансплантацию костного мозга с вероятностью долговременного выживания 35–65%. Взрослые редко болеют ОЛЛ. Длительной ремиссии (более 5 лет) удаётся достичь в 15–25% случаев. Острый миелобластный лейкоз Полной ремиссии при большинстве вариантов острого миелобластного лейкоза удаётся добиться у 60–70% больных. При различных постиндукционных схемах средняя длительность ремиссии — 12–15 мес; у 25–35% больных нет рецидивов в течение 24 мес, часть из них достигает стойкой ремиссии («излечивается»). Прогностически неблагоприятные признаки Мужской пол и возраст более 50 лет. Мегакариобластный и эритробластный тип острого миелоидного лейкоза. Ph-позитивный острый лимфолейкоз. В дебюте заболевания — гиперлейкоцитоз свыше 50 Возможные исходы острых лейкозов • Смерть больного. Причины смерти при острых лейкозах — нейролейкемия, геморрагический синдром (ДВС), почечная недостаточность (при синдроме лизиса опухолевых клеток), сердечная недостаточность, инфекционные осложнения (сепсис), острая надпочечниковая недостаточность (вследствие гипотрофии коры надпочечников при отмене длительно применяемых глюкокортикоидов). • Ремиссия заболевания в течение 5 лет от завершения курса терапии. • Выздоровление. О полном выздоровлении говорят при отсутствии рецидива в течение 5 лет после завершения полного курса терапии. • Рецидив заболевания. Может быть ранним и поздним: ранний — во время лечения; поздний — после проведённого лечения. ХРОНИЧЕСКИЕ ЛЕЙКОЗЫ Хронический лимфолейкоз Большинство (85%) хронических лимфоидных лейкозов (ХЛЛ) возникает из предшественников B–лимфоцитов. Хронический В-клеточный лимфолейкоз (B-ХЛЛ) — опухоль из СD5+-позитивных В-клеток, первично поражающая костный мозг. Заболеваемость варьирует в различных географических регионах и этнических группах, но болеют в основном пожилые; В-ХЛЛ составляет около 25% всех лейкозов, встречающихся в пожилом возрасте. Детская заболеваемость казуистична. У молодых заболевание протекает с явными признаками злокачественности, молодой возраст считается одним из признаков плохого прогноза. Мужчины болеют вдвое чаще женщин. Патогенез На уровне предшественника В-клетки происходит хромосомная аберрация, приводящая к трисомии хромосомы 12 либо к структурным нарушениям хромосом 6, 11, 13 или 14. Патологические клетки дифференцируются до уровня рециркулирующих В-клеток или B-клеток памяти. Их нормальные клеточные аналоги — длительноживущие, иммунологически ареактивные, митотически пассивные В-клетки Тнезависимого пути дифференцировки и B-клетки памяти соответственно. Последующие деления генетически нестабильных лимфоцитов могут привести к появлению новых мутаций и, соответственно, новых биологических свойств, т.е. субклонов лейкозных клеток. Проявления: Стадия 0 ХЛЛ характеризуется лишь лимфоцитозом. Прогноз благоприятный (средняя продолжительность жизни — 10–12 лет). Стадия I манифестируется лимфаденопатией (увеличиваются лимфатические узлы верхней половины тела, в основном шейные, надключичные и подмышечные; они мягкой консистенции). Стадия II проявляется спленомегалией. Прогноз неблагоприятный (пациенты обычно живут 4–7 лет). Стадия III сопровождается развитием аутоиммунной гемолитической анемии. Стадия IV характеризуется возникновением тромбоцитопении (иммуноаутоагрессивного генеза). Длительность жизни пациентов составляет менее 18 мес. Кроме того, у пациентов, как правтло наблюдаются: • Анемический и геморрагический синдромы (обусловлены поражением костного мозга и появлением АТ к эритроцитам и тромбоцитам). • Инфекционные осложнения (вызваны гипогаммаглобулинемией, нарушениями клеточного звена иммунитета, миграции и хемотаксиса гранулоцитов. Часто отмечаются бактериальные, грибковые и вирусные инфекции). • Аллергические реакции немедленного типа (наиболее часто на вакцинации, укусы комаров). ХРОНИЧЕСКИЙ МИЕЛОЛЕЙКОЗ Хронический миелолейкоз (ХМЛ) — миелоидная опухоль, развивающаяся из полипотентной клетки-предшественницы. Её пролиферация и дифференцировка приводит к расширению ростков кроветворения, представленных (в отличие от острых лейкозов) преимущественно зрелыми и промежуточными формами лейкозных клеток. В большинстве случаев закономерным исходом болезни является бластный криз, характеризующийся появлением большого количества бластных клеток и рефрактерностью к терапии, и заканчивающийся летально. Распространённость ХМЛ — наиболее распространённый из всех лейкозов. На него приходится 20% случаев гемобластозов у взрослых и 5% — у детей. Этиология и патогенез На уровне клетки-предшественницы происходит транслокация t(9;22), что приводит к появлению так называемой «филадельфийской» хромосомы и экспресии мутантного гена bcr-abl, кодирующего белок p210, обладающий свойствами тирозинкиназы. Экспансия Ph-позитивных клеток в костном мозге, периферической крови и экстрамедуллярных областях объяснима не столько их высокой пролиферативной активностью, сколько расширением пула гранулоцитарных предшественников, утерявших чувствительность к регуляторным стимулам и изменениям микроокружения. Это приводит к их диссеминации, нарушению продукции цитокинов и подавлению нормального гемопоэза. Период полужизни гранулоцита при ХМЛ превышает таковой нормального гранулоцита в 10 раз. Транслокация t(9;22) является диагностической для ХМЛ. При её отсутствии заболевание, характеризующееся клиническими, морфологическими и цитохимическими признаками ХМЛ, определяется как атипичный хронический миелолейкоз. Стадии и диагностика Развёрнутая стадия Диагноз устанаиливатся на основании наличия «немотивированной» природы нейтрофильного лейкоцитоза со сдвигом формулы до миелоцитов и промиелоцитов, существенно повышенного соотношения «лейкоциты/эритроциты» в костном мозге, «филадельфийской» хромосомы в гранулоцитах крови и клетках костного мозга. В трепанате костного мозга уже в этот период, как правило, наблюдается почти полное вытеснение жира миелоидной тканью. Терминальная стадия . Для этой стадии характерны появление и интенсивное нарастание признаков подавления нормальных ростков кроветворения — анемия, тромбоцитопения с геморрагическим синдромом; гранулоцитопения, осложняющаяся инфекцией; некрозами слизистых оболочек. Важнейшим гематологическим признаком терминальной стадии хронического миелолейкоза является бластный криз — увеличение содержания бластных клеток в костном мозге и крови (сначала миелобластов, затем — морфологически недифференцируемых бластов). На терминальной стадии более чем в 80% случаев появляются анэуплоидные клоны клеток — кроветворные клетки, содержащие ненормальное число хромосом. Длительность жизни больных на этой стадии чаще не превышает 6–12 мес. ЛЕЙКЕМОИДНЫЕ РЕАКЦИИ Лейкемоидные реакции — состояния, характеризующиеся изменениями в крови, органах гемопоэза и организме в целом, сходные с теми, которые наблюдаются при гемобластозах, главным образом — при лейкозах. Своё название — «лейкемоидные» — эти реакции получили в связи с тем, что изменения в гемопоэтической ткани и в периферической крови напоминают изменения при лейкозах. Однако, лейкемоидные реакции не трансформируются в тот лейкоз, с которым они сходны гематологически. Отличия между лейкемоидными реакциями и лейкозами приведены в табл. 21–5. Таблица 21–5. Отличия лейкемоидных реакций от лейкоза
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||