Патфиз ч.1. Патфиз ч. Гл. 1 Введение в предмет Гл. 2 Общая нозология
 Скачать 9.21 Mb. Скачать 9.21 Mb.
|

|
В зависимости от функционального класса изменённого полипептида (белки структурные, ферменты, рецепторы, трансмембранные переносчики и т.д.) делаются попытки классифицировать моногенные болезни. На настоящий момент очевидно, что мутантные гены, кодирующие ферменты, приводят к развитию энзимопатий, наиболее часто встречающихся моногенных болезней. Для любого моногенного заболевания существенной характеристикой является тип наследования: аутосомно–доминантный, аутосомно–рецессивный, сцепленный с хромосомой X (доминантный и рецессивный), голандрический (сцепленный с хромосомой Y) и митохондриальный (рис. 3–7). Клинические проявления моногенных болезней зависят от пенетрантности и экспрессивности. Пенетрантность оценивается по проценту переносчиков, у которых обнаруживаются фенотипические проявления, вызванные экспрессией мутантного гена: если выявляется хотя бы один признак или симптом болезни, то считается, что фенотипические проявления гена имеются. Пенетрантность во многом зависит от воздействия факторов внешней среды. Например, у индивидуумов–гетерозигот с дефицитом глюкозо-6-фосфат дегидрогеназы гемолиз эритроцитов происходит только при действии оксидантов. Следует помнить также, что при некоторых заболеваниях их симптомы появляются лишь в зрелом возрасте. Поэтому говорить о пенетрантности мутантного гена можно при достижении индивидуумом определенного возраста. Экспрессивность описывает степень влияния гена на фенотип. Экспрессивность описывается видом и тяжестью симптомов и зависит от возраста, при котором возникло заболевание. Термины «доминантный» и «рецессивный» характеризуют фенотип, которой определяется аллельной парой. При заболеваниях с рецессивным типом наследования фенотип гетерозиготы может не отличаться от нормы (т.е., иметь слабые проявления заболевания или не иметь их). При заболеваниях с доминантным типом наследования пациенты в гетерозиготном состоянии имеют практически ту же картину заболевания, что и больные в гомозиготном состоянии. Однако, проявления заболевания у гомозигот более тяжелые, чем у гетерозигот. В связи с этим, появилось рабочее понятие о полудоминантном или частично доминантном типе наследования заболеваний.  Рис. 3–7. Виды генных болезней. АУТОСОМНОДОМИНАНТНЫЙ ТИП НАСЛЕДОВАНИЯ Примеры заболеваний: синдром Марфана, гемоглобиноз M, хорея Хантингтона, полипоз толстой кишки, семейная гиперхолестеринемия, нейрофиброматоз, полидактилия. Родословная с аутосомнодоминантным типом наследования патологии — синдрома Марфана в пяти поколениях — представлена на рис. 3–8. 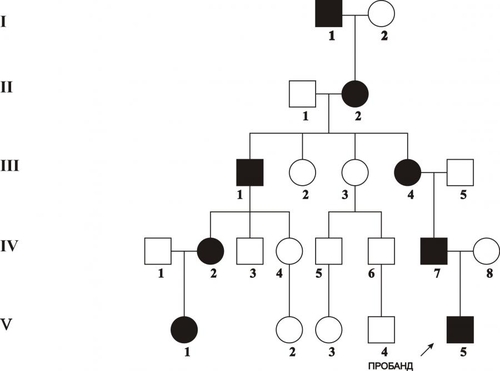 Рис. 3–8. Родословная с аутосомнодоминантным типом наследования заболевания. Кружок — пол женский, квадрат — пол мужской, тёмный кружок и/или квадрат — больной. Особенности наследования Один из родителей пациента, как правило, болен. Исключением являются случаи, когда заболевание является результатом впервые возникшей мутации. Выраженность и количество проявлений зависят от действия факторов внешней среды. Частота патологии у лиц мужского и женского пола одинаковая. В каждом поколении имеются больные (так называемый вертикальный характер распределения болезни). Вероятность рождения больного ребёнка равна 50% (независимо от пола ребёнка и количества родов). Непоражённые члены семьи, как правило, имеют здоровых потомков (поскольку не имеют мутантного гена). АУТОСОМНОРЕЦЕССИВНЫЙ ТИП НАСЛЕДОВАНИЯ Примеры заболеваний: фенилкетонурия, адреногенитальный синдром, кожноглазной альбинизм, галактоземия, гликогенозы, гиперлипопротеинемии, муковисцидоз. Родословная с аутосомнорецессивным типом наследования муковисцидоза в четырёх поколениях представлена на рис. 3–9. 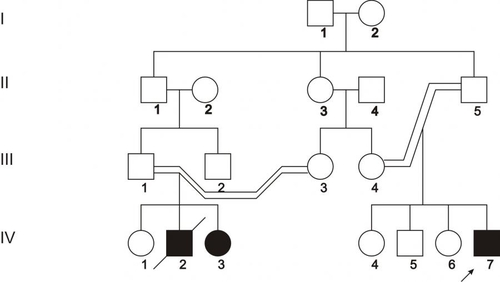 Рис. 3–9. Родословная с аутосомнорецессивным типом наследования заболевания. Кружок — пол женский, квадрат — пол мужской, тёмный кружок и/или квадрат — больной, наискось перечёркнутый тёмный кружок и/или квадрат — умерший больной. Особенности наследования Родители больного, как правило, здоровы. Это заболевание может обнаруживаться у других родственников, например у двоюродных или троюродных братьев (сестёр) больного. В гомозиготном состоянии проявления болезни более униформны, в связи с высокой пенетрантностью. Симптомы болезни обычно выявляются уже в детском возрасте. Частота патологии у лиц мужского и женского пола равная. В родословной патология проявляется «по горизонтали», часто у сибсов. Заболевание отсутствует у единокровных (дети одного отца от разных матерей) и единоутробных (дети одной матери от разных отцов) братьев и сестёр. Появление аутосомнорецессивной патологии более вероятно при кровнородственных браках за счёт большей вероятности встречи двух супругов, гетерозиготных по одному и тому же патологическому аллелю, полученному от их общего предка. Чем больше степень родства супругов, тем эта вероятность выше. СЦЕПЛЕННОЕ С ХРОМОСОМОЙ X ДОМИНАНТНОЕ НАСЛЕДОВАНИЕ Примеры заболеваний: одна из форм гипофосфатемии — витамин Dрезистентный рахит, болезнь Шарко–Мари–Тута Xсцепленная доминантная, ротолицепальцевой синдром типа I. Родословная с доминантным X–сцепленным типом наследования витамин D–резистентного рахита в четырёх поколениях представлена на рис. 3–10. 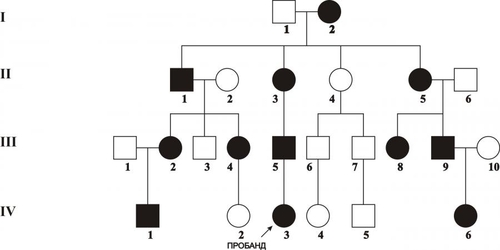 Рис. 3–10. Родословная с доминантным Xсцепленным типом наследования заболевания. Кружок — пол женский, квадрат — пол мужской, тёмный кружок и/или квадрат — больной. Особенности наследования Поражение лиц мужского и женского пола, но женщин — в 2 раза чаще. При этом у мужчин отмечается более тяжёлое течение заболевания. Передача больным мужчиной патологического аллеля всем дочерям и только дочерям, но не сыновьям. Сыновья получают от отца хромосому Y. Передача больной женщиной заболевания и сыновьям, и дочерям с равной вероятностью. СЦЕПЛЕННОЕ С ХРОМОСОМОЙ X РЕЦЕССИВНОЕ НАСЛЕДОВАНИЕ Примеры заболеваний: гемофилия A, гемофилия B, Xсцепленная рецессивная болезнь Шарко–Мари–Тута, дальтонизм, мышечная дистрофия Дюшенна–Беккера, синдром Калльмана, болезнь Хантера (мукополисахаридоз типа II), гипогаммаглобулинемия брутоновского типа. Родословная с рецессивным X–сцепленным типом наследования гемофилии А в четырёх поколениях представлена на рис. 3–11. 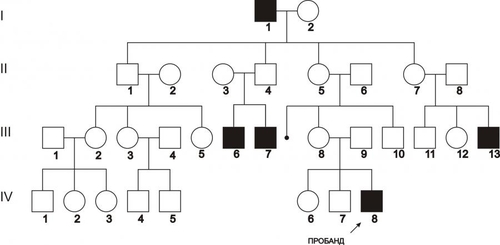 Рис. 3–11. Родословная с рецессивным Xсцепленным типом наследования заболевания. Кружок — пол женский, квадрат — пол мужской, тёмный кружок и/или квадрат — больной. Признаки заболевания Больные рождаются в браке фенотипически здоровых родителей. Заболевание наблюдается почти исключительно у лиц мужского пола. Матери больных — облигатные носительницы патологического гена. Сын никогда не наследует заболевание от отца. У носительницы мутантного гена вероятность рождения больного ребёнка равна 25% (независимо от пола новорождённого), вероятность рождения больного мальчика равна 50%. ГОЛАНДРИЧЕСКИЙ, ИЛИ СЦЕПЛЕННЫЙ С ХРОМОСОМОЙ Y, ТИП НАСЛЕДОВАНИЯ Гены, ответственные за развитие патологического признака, локализованы в хромосоме Y. Примеры признаков: гипертрихоз ушных раковин, избыточный рост волос на средних фалангах пальцев кистей, азооспермия. Родословная с Y–сцепленным типом наследования избыточного оволосения ушных раковин в четырёх поколениях представлена на рис. 3–12. 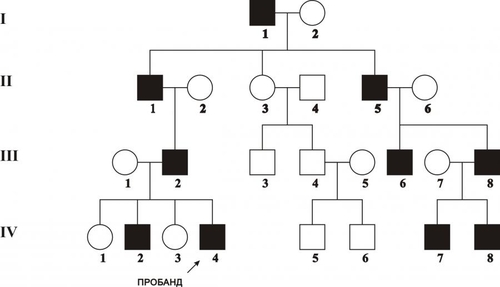 Рис. 3–12. Родословная с Yсцепленным (голандрическим) типом наследования. Кружок — пол женский, квадрат — пол мужской, тёмный кружок и/или квадрат — больной. Особенности наследования Передача признака от отца всем сыновьям и только сыновьям. Дочери никогда не наследуют признак от отца. «Вертикальный» характер наследования признака. Вероятность наследования для лиц мужского пола равна 100%. МИТОХОНДРИАЛЬНОЕ НАСЛЕДОВАНИЕ Геном митохондрий полностью секвенирован. Он содержит 16 569 пар оснований и кодирует две рибосомные РНК (12S и 16S), 22 транспортные РНК и 13 полипептидов — субъединиц ферментативных комплексов окислительного фосфорилирования. Другие 66 субъединиц дыхательной цепи кодируются в ядре. Примеры заболеваний (митохондриальные болезни): атрофия зрительного нерва Лебера, синдромы Лея (митохондриальная миоэнцефалопатия), MERRF (миоклоническая эпилепсия), кардиомиопатия дилатационная семейная. Родословная с митохондриальным типом наследования — атрофии зрительного нерва Лебера в четырёх поколениях — представлена на рис. 3–13. 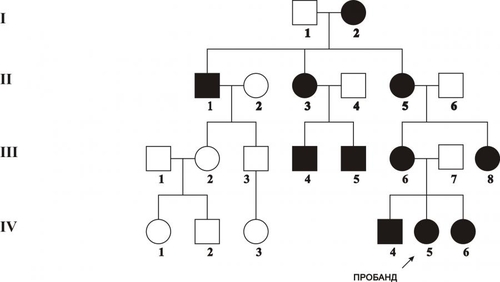 Рис. 3–13. Родословная с митохондриальным типом наследования заболевания. Кружок — пол женский, квадрат — пол мужской, тёмный кружок и/или квадрат — больной. Особенности наследования Наличие патологии у всех детей больной матери. Рождение здоровых детей у больного отца и здоровой матери. Указанные особенности объясняются тем, что митохондрии наследуются от матери. Доля отцовского митохондриального генома в зиготе составляет ДНК от 0 до 4 митохондрий, а материнского генома — ДНК примерно 2500 митохондрий. К тому же после оплодотворения репликация отцовской ДНК блокируется. ПРИМЕРЫ МОНОГЕННЫХ ЗАБОЛЕВАНИЙ ФЕНИЛКЕТОНУРИЯ Все формы фенилкетонурии — последствия недостаточности группы ферментов. Их гены транскрибируются в гепатоцитах и наследуются по аутосомнорецессивному типу. Наиболее частая форма фенилкетонурии возникает при мутациях гена фенилаланин 4монооксигеназы (фенилаланин 4-гидроксилаза, фенилаланиназа). Самый распространённый тип мутаций — однонуклеотидные замены (миссенс, нонсенсмутации и мутации в сайтах сплайсинга). Ведущее патогенетическое звено: гиперфенилаланинемия с накоплением в тканях токсических продуктов (фенилпировиноградной, фенилуксусной, фенилмолочной и других кетокислот). Это ведёт к поражению ЦНС, нарушению функции печени, обмена белков, липо- и гликопротеинов, метаболизма гормонов. Проявления: повышенная возбудимость и гипертонус мышц, гиперрефлексия и судороги, признаки аллергического дерматита, гипопигментация кожи, волос, радужки; «мышиный» запах мочи и пота, задержка психомоторного развития. У нелеченых детей формируется микроцефалия и умственная отсталость, откуда и другое название заболевания — фенилпируватная олигофрения. Терапия: коррекция с помощью диетотерапии (исключение или снижение содержания в пище фенилаланина). Диету необходимо соблюдать с момента установления диагноза (первые сутки после рождения) и контролировать содержание фенилаланина в крови не менее 8–10 лет. АЛЬБИНИЗМ КОЖНОГЛАЗНОГО ТИПА Существует несколько форм кожно–глазного альбинизма. Тип IА кожно–глазного альбинизма характеризуется недостаточностью тирозиназы (у гомозигот активность фермента отсутствует полностью); частота примерно 1:10 000; наследование аутосомно–рецессивное. Проявления: тотальная депигментация кожи, волос и радужки глаз, полное отсутствие какихлибо пигментных пятен, краснорозовая, не загорающая кожа, предрасположенная к малигнизации; нистагм и светобоязнь, снижение остроты зрения, не улучшающегося с возрастом, нарушение бинокулярного зрения. ГЕМОФИЛИЯ А (СМ. СТАТЬЮ «ГЕМОФИЛИЯ» В ПРИЛОЖЕНИИ «СПРАВОЧНИК ТЕРМИНОВ» НА КОМПАКТ-ДИСКЕ) Синдром Марфана Частота синдрома Марфана находится в диапазоне 1:10 000–15 000, наследуется он аутосомнодоминантному типу. Инициальное патогенетическое звено — мутация гена фибриллина (FBN1). Идентифицировано около 70 мутаций (преимущественно миссенстипа). Мутации различных экзонов гена FBN1 вызывают различные изменения фенотипа, от умеренно выраженных (субклинических) до тяжёлых. Проявления: признаки множественных дефектов соединительной ткани (поскольку фибриллин широко представлен в матриксе соединительной ткани кожи, лёгких, сосудов, почек, мышц, хрящей, сухожилий, связок), поражения скелета (высокий рост, диспропорционально длинные конечности, арахнодактилия), поражения сердечнососудистой системы (расслаивающая аневризма аорты, пролапс митрального клапана), поражение глаз (вывих или подвывих хрусталика, дрожание радужки). ГЕМОГЛОБИНОПАТИЯ S Гемоглобинопатия S (аутосомнорецессивное наследование) распространена в странах так называемого малярийного пояса Земли. Это объясняется тем, что гетерозиготы по HbS резистентны к тропической малярии. В частности, носители HbS распространены в Закавказье и Средней Азии, в России максимальная частота гетерозиготных носителей HbS отмечена в Дагестане. Возникновение HbS происходит при замещении одного основания в 6м триплете (миссенсмутация) Гетерозиготные носители HbS в обычных условиях здоровы, но при пониженном pO2 (кессонные работы, условия высокогорья и т.д.) или при гипоксемии (ВПР сердца, дыхательная недостаточность, длительный наркоз и т.п.) развивается гемолитическая анемия. Гомозиготы страдают тяжёлой гемолитической анемией с 4–6месячного возраста. В результате капиллярного или венозного тромбоза серповидными эритроцитами развиваются трофические язвы (часто на голени), боли в животе, поражение сердца, глаз. Характерны поражения костносуставной системы, гепатоспленомегалия. МУКОВИСЦИДОЗ Муковисцидоз — множественное поражение экзокринных желёз, сопровождающееся накоплением и выделением ими вязких секретов. Среди новорождённых частота составляет примерно 1:1500–1:2000. Кистозный фиброз является одним из самых распространённых моногенных заболеваний в Европе. Наследуется муковисцидоз по аутосомнорецессивному типу. Известно более 130 аллелей; наиболее частая мутация delF508, приводящая к отсутствию фенилаланина в 508-м положении трансмембранного регуляторного белка. В зависимости от типа мутаций и их локализации функция гена может быть полностью или частично нарушена. При мутации нарушается регуляция переноса Cl– через мембраны эпителиальных клеток (транспорт Cl– тормозится, а Na+ усиливается). Болезнь характеризуется закрытием протоков желёз вязким секретом, который образуется в связи с повышенной резорбцией Na+ клетками протоков экзокринных желёз. Нередко в протоках образуются кисты и развивается воспаление. При хроническом течении в железах развивается избыток соединительной ткани (склероз). У новорождённых нередко выявляется непроходимость кишечника (мекониальный илеус). У детей наиболее часто развивается лёгочная или лёгочнокишечная форма. Они проявляются повторными бронхитами, пневмониями, эмфиземой лёгких, а также нарушениями полостного и пристеночного пищеварения, вплоть до развития синдрома мальабсорбции (синдром нарушенного всасывания). При длительном течении развиваются дыхательная недостаточность, цирроз печени, портальная гипертензия, нередко приводящие к смерти. ХРОМОСОМНЫЕ БОЛЕЗНИ Хромосомные болезни выявляются у новорождённых с частотой 6:1000. Инициальное звено патогенеза — геномная или хромосомная мутация. Хромосомный дисбаланс приводит к остановке либо нарушению эмбрионального развития, в том числе ранних этапов органогенеза. В результате формируются множественные ВПР. Тяжесть нарушений обычно коррелирует со степенью хромосомного дисбаланса: чем больше хромосомного материала вовлечено в аберрацию, тем раньше проявляется хромосомный дисбаланс в онтогенезе, тем значительнее нарушения физического и психического развития индивида. Как правило потеря хромосомы или ее части приводит к более тяжелым клиническим последствиям, чем присоединение хромосомы или ее части. Хромосомные болезни классифицируют (рис. 3–14) по критериям изменения структуры и числа хромосом, а также в зависимости от типа клеток (половые или соматические). 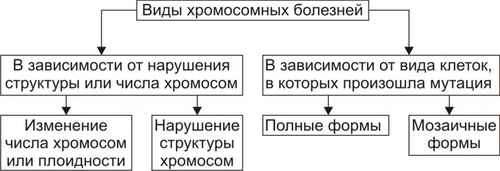  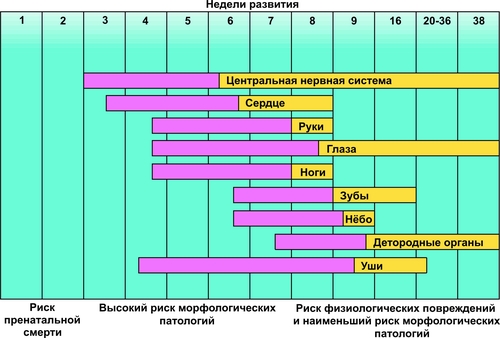 Рис. 3–14. Виды хромосомных болезней. Большинство геномных мутаций (полиплоидии, трисомии по крупным хромосомам, моносомии по аутосомам) летальны. Насчитываются сотни болезней, вызванных нарушением структуры хромосом в результате делеции, дупликации, инверсии или транслокации их отдельных участков. Их клиническая картина и тяжесть определяются характером перестройки, величиной вовлеченных фрагментов и их функциональной значимостью. Мутации в гаметах приводят к развитию так называемых полных форм хромосомных болезней, когда изменения кариотипа выявляются во всех клетках организма. Мутации в соматических клетках на ранних этапах эмбриогенеза приводят к развитию мозаицизма: часть клеток организма имеет нормальный кариотип, а другая часть — аномальный. Это вызывает так называемые мозаичные формы хромосомных болезней. Варианты мозаичных организмов могут быть самыми разнообразными: не только из двух, но из трёх и более клонов клеток с разными их количественными соотношениями. Фенотипические отклонения от нормы зависят от доли клеток различных типов, т.е. от стадии развития, на которой произошла мутация. Для хромосомных болезней характерно нарушение репродуктивной функции. |