Патфиз ч.1. Патфиз ч. Гл. 1 Введение в предмет Гл. 2 Общая нозология
 Скачать 9.21 Mb. Скачать 9.21 Mb.
|

|
Синдром Кляйнфелтера Частота: 2–2,5 на 1000 новорождённых мальчиков. В кариотипе разнообразные цитогенетические варианты (47,XXY; 48,XXXY; 49,XXXXY и др.), но чаще встречается классический вариант 47,XXY. Проявления: высокий рост, непропорционально длинные конечности, отложение жира по женскому типу, евнухоидное телосложение, скудное оволосение, гинекомастия, гипогенитализм, бесплодие (в результате нарушения сперматогенеза, снижения продукции тестостерона и увеличения продукции женских половых гормонов), снижение интеллекта (чем больше в кариотипе добавочных хромосом, тем более выражено). Лечение описанной патологии мужскими половыми гормонами направлено на коррекцию вторичных половых признаков. Однако, и после терапии больные остаются бесплодными. СИНДРОМЫ ПОЛИСОМИИ Х Трисомия Х. Наиболее частым синдромом из группы полисомий X является синдром трисомии Х (47,XXX): частота 1:1000 новорождённых девочек, кариотип 47,XXX; пол — женский, фенотип женский; как правило, физическое и психическое развитие у женщин с этим синдромом не имеет отклонений от нормы. Тетра и пентасомии Х: высокий рост, телосложение по мужскому типу, челюстнолицевые дисплазии (эпикант, гипертелоризм, готическое нёбо и т.д.), нарушения менструального цикла, бесплодие, преждевременный климакс, снижение интеллекта различных степеней (отмечено у двух третей больных), нередки психических заболевания (шизофрения, маниакальнодепрессивный психоз, эпилепсия). Синдром Шерешевского–Тёрнера Частота синдрома: 1:3000 новорождённых девочек; кариотип: 45,Х0, но встречаются и другие варианты (например, изохромосома длинного плеча X — Xqi, делеция короткого плеча — Xp, делеция длинного плеча —Xq). Проявления: низкий рост, короткая шея с избытком кожи или крыловидной складкой, широкая, часто деформированная грудная клетка, деформация локтевых суставов, недоразвитие первичных и вторичных половых признаков, бесплодие. O новорождённых почти во всех случаях находят лимфатический отёк кистей и стоп. Раннее лечение женскими половыми гормонами может оказаться эффективным. БОЛЕЗНИ С НАСЛЕДСТВЕННЫМ ПРЕДРАСПОЛОЖЕНИЕМ Болезни с наследственным предрасположением называют также многофакторными, так как их возникновение определяется взаимодействием наследственных факторов и разнообразных факторов внешней среды. В основе предрасположенности к болезням находится генетическое разнообразие (генетический полиморфизм) популяций по ферментам, структурным, транспортным белкам, антигенным системам и т.д. Количественная оценка вклада наследственного и средового факторов при возникновении болезней с наследственным предрасположением рассчитывается по специальным формулам. Наиболее распространён расчёт коэффициента наследуемости (Н) и роли средовых факторов (Е, от англ. environment — окружающая среда) по формуле, предложенной Хольцингером: где 100 — Кдз; Кмз —% конкордантных по данному признаку (болезни) в данной выборке монозиготных близнецов (по отношению ко всей их популяции); Кдз —% конкордантности по данному признаку (болезни) в данной выборке дизигот по отношению ко всей популяции близнецов. С учётом коэффициента Хольцингера можно рассчитать роль факторов окружающей среды в возникновении данной патологии — коэффициент E: Частота болезней с наследственным предрасположением — более 90% всех неинфекционных форм патологий. К болезням с наследственным предрасположением относятся ИБС, гипертоническая болезнь, бронхиальная астма, психические заболевания, СД, ревматические болезни, язвенная болезнь желудка, ВПР и многие другие. ВИДЫ МНОГОФАКТОРНЫХ БОЛЕЗНЕЙ Болезни с наследственным предрасположением классифицируют — в зависимости от числа генов, определяющих предрасположенность, — на моногенные и полигенные. МОНОГЕННЫЕ ЗАБОЛЕВАНИЯ Моногенные болезни с наследственным предрасположением детерминируются одним мутантным геном и возникают при действии конкретного (часто специфического) и обязательного фактора внешней среды. К таким разрешающим факторам относятся загрязнение среды (химическими соединениями, пылевыми частицами), пищевые вещества и добавки, ЛС. Примеры: • непереносимость лактозы: при мутантной форме гена лактазы употребление молока приводит к развитию кишечного дискомфорта и поноса; • у гомозигот по мутантному аллелю гена, регулирующего реакцию ацетилирования изониазида (противотуберкулёзного препарата), стандартная доза этого препарата вызывает поражение периферических нервов, которое проявляется невритами. ПОЛИГЕННЫЕ БОЛЕЗНИ Предрасположенность к развитию полигенных болезней детерминируется взаимодействием нормальных и/или изменённых (мутировавших) генов. Каждый из них по отдельности не приводит к развитию заболевания. Индивид с такой комбинацией генов достигает «порога возникновения» болезни и заболевает. Этот порог может быть преодолён под действием определённого фактора окружающей среды. ХАРАКТЕРИСТИКА МНОГОФАКТОРНЫХ БОЛЕЗНЕЙ • Наследование многофакторных болезней не соответствует менделевским закономерностям. • Патогенез болезней с наследственным предрасположением зависит от «удельного вклада» генетических и средовых факторов. Эта зависимость различна как для разных заболеваний, так и для каждого человека. Именно полиморфизм создаёт основу для предрасположенности организма к той или иной патологии. • Многофакторные болезни возникают в результате взаимодействия предрасположенного организма с комплексом неблагоприятных факторов внешней среды. Чем выше генетическая предрасположенность организма (т.е. чем ближе к «порогу возникновения» болезни он находится), тем менее интенсивным и длительным должно быть воздействие средового фактора для запуска патологического процесса, заболевания или состояния. • Для многофакторных болезней характерно наличие большого числа клинических вариантов. Они образуют ряд переходных состояний: от минимальных, клинически стёртых форм до тяжёлых проявлений. • При болезнях с наследственным предрасположением наблюдается более высокая конкордантность по заболеванию у монозиготных близнецов в сравнении с дизиготными. ВРОЖДЁННЫЕ ПОРОКИ РАЗВИТИЯ Врождённые пороки развития (ВПР), включая аномалии развития, дисплазии и стигмы дизэмбриогенеза, а также причины их появления изучает тератология. Механизм формирования ВПР в ходе внутриутробного развития обозначается как тератогенез, а термин «тератоген» означает фактор, вызвавший ВПР. Большинство ВПР обусловлено воздействием факторов внешней среды, генетическими дефектами или их сочетанием. В ряде случаев не удаётся установить причину ВПР (спорадические заболевания). Число новорождённых с ВПР составляет 2–3% общего количества родившихся живыми детей. Причины ВПР многочисленны: вирусная инфекция (краснуха, цитомегаловирусная и герпетическая инфекции), токсоплазмоз, сифилис, радиация, ЛС, наркотические вещества, химические факторы окружающей среды, болезни матери и т.д. Восприимчивость к действию тератогенов зависит от стадии развития. Риск возникновения ВПР особенно велик в периоды эмбриогенеза и органогенеза. ПРИЧИНЫ ВПР ТЕРАТОГЕННЫЕ ВОЗДЕЙСТВИЯ Тератогенные факторы — те средовые факторы, которые нарушают развитие эмбриона и/или плода, воздействуя на эмбрион и/или плод в течение беременности. Около 10% всех ВПР обусловлено воздействием факторов внешней среды. Эффект тератогенов обусловлен влиянием на гисто- и органогенез, рост и развитие плода. ГЕНЕТИЧЕСКИЕ НАРУШЕНИЯ Генетические факторы могут приводить как к единичным ВПР, так и к развитию многообразных синдромов. СПОРАДИЧЕСКИЕ ЗАБОЛЕВАНИЯ Спорадические заболевания часто бывают следствием нарушения эмбрионального развития или патологического течения беременности (например, при окклюзии кровеносных сосудов). Некоторые врождённые аномалии могут возникать в результате спонтанной доминантной мутации ( ФАКТОРЫ РИСКА ВПР Факторы риска развития ВПР подразделяют на эндогенные и экзогенные (средовые). ЭНДОГЕННЫЕ ФАКТОРЫ РИСКА Среди эндогенных факторов риска ВПР выделяют мутагены, эндокринные и метаболические заболевания матери, аномалии половых клеток, возраст родителей. • Мутагены могут вызвать изменения генетического аппарата, на долю генных и хромосомных мутаций приходится более 30% всех ВПР. † Генные мутации являются причиной около 20% от всех ВПР (например, расщелины губы и нёба как одно из клинических проявлений синдрома Ван дер Вуда). † Хромосомные мутации обусловливают развитие примерно 10% ВПР (например, пороки сердца при синдроме Дауна). • Эндокринные заболевания и метаболические расстройства в организме матери нарушают развитие органов плода или приводят к самопроизвольным абортам. Наиболее часто ВПР наблюдаются при СД, вирилизирующих опухолях половых желёз и коры надпочечников, фенилкетонурии. • При аномалиях половых клеток (результат нарушения спермато и/или овогенеза) возникают анеуплоидии и триплоидии. • Возраст родителей. † Возраст отца. Установлена прямая зависимость между нарастанием частоты некоторых ВПР (например, расщелины губы и нёба) и аутосомнодоминантных наследственных заболеваний (например, ахондроплазии) и увеличением возраста отца. † Возраст матери. ВПР дыхательной системы чаще отмечаются у юных матерей. У матерей старших возрастных групп увеличена частота рождения детей с геномными мутациями (классический пример: значительное увеличение трисомий, в том числе синдром Дауна). ЭКЗОГЕННЫЕ ФАКТОРЫ РИСКА На долю средовых факторов риска приходится около 10% всех ВПР. Природа экзогенных факторов риска может быть физической, химической, биологической и сочетанной. • Физические воздействия: радиационные, вибрационные, шумовые, температурные, механические. Из механических факторов большое клиническое значение имеют амниотические сращения, маловодие и миомы матки. † Амниотические сращения (тяжи Симонара) могут приводить к перетяжкам на конечностях, вызывая гипоплазию их дистальных отделов или ампутацию. † Маловодие может обусловить развитие ВПР конечностей, гипоплазии нижней челюсти и др. † Крупные миомы препятствуют нормальному росту и развитию эмбриона или плода. • Химические агенты † Некоторые ЛС (например, антиконвульсант гидантоин вызывает развитие расщелины губы и нёба, микроцефалию, гипоплазию ногтей и концевых фаланг пальцев, деформацию носа; транквилизатор талидомид может вызвать ВПР верхних и нижних конечностей [вплоть до амелии], расщелины губы и нёба) † Химические вещества, применяемые в быту и в промышленности (например, продукты метаболизма этанола могут привести к алкогольной эмбрио или фетопатии; бензин, бензол, фенол, соли тяжёлых металлов обладают эмбриотоксическими свойствами). • Биологические (например, вирусы краснухи и цитомегаловирусной инфекции; вирус краснухи вызывает поражение ЦНС, ВПР органов зрения и слуха). • Сочетанные воздействия (как результат совместного влияния генетических и средовых факторов) могут привести к развитию ВПР. При этом ни один из них отдельно не является причиной ВПР. Их доля среди всех причин ВПР составляет примерно 50%. СТАДИЯ ВНУТРИУТРОБНОГО РАЗВИТИЯ ПЛОДА Степень воздействия на эмбрион зависит от срока беременности на момент воздействия: 2-4 нед. после оплодотворения: плод либо развивается нормально, либо гибнет; 4-12 нед.: возникают микроцефалия, умственная отсталость, катаракта, задержка роста, микрофтальмия; 12-16 нед: развивается умственная отсталость или задержка роста; после 20 нед: повреждение волосяных фолликулов, поражение кожи и слизистых оболочек, угнетение красного костного мозга. Критические сроки развития возможных пороков развития по системам органов. [по 4]. ТИПЫ ВПР В зависимости от срока беременности при воздействия повреждающих факторов выделяют гаметопатии, бластопатии, эмбриопатии и фетопатии (рис. 3–15). 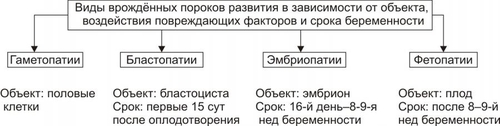 Рис. 3–15. Виды врождённых пороков. Гаметопатии — результат воздействия на половые клетки (например, все ВПР, в основе которых находятся мутации в половых клетках). Бластопатии — следствие поражения бластоцисты — зародыша первых 15 сут после оплодотворения (до завершения формирования зародышевых листков). Результатом бластопатий являются, например, двойниковые пороки (сросшиеся близнецы), циклопия (наличие одного или двух слившихся глазных яблок в единственной орбите по срединной линии лица). Эмбриопатии — результат воздействия тератогенного фактора на эмбрион в период с 16-го дня до 8–9 недели беременности. К этой группе относятся талидомидные, диабетические, алкогольные и некоторые медикаментозные эмбриопатии, а также ВПР, развившиеся под влиянием вируса краснухи. Фетопатии — следствие повреждения плода от 9-й недели до момента рождения. К фетопатиям относятся, например, крипторхизм, открытый боталлов проток или пренатальная гипоплазия какого–либо органа или плода в целом. МЕХАНИЗМЫ РАЗВИТИЯ Механизмы развития ВПР заключаются в искажениях межмолекулярных и межклеточных взаимодействий, а также в нарушениях морфогенетических процессов. Расстройства межмолекулярных и межклеточных взаимодействий приводят к нарушениям синтеза биологически активных веществ (гормонов, цитокинов и др.), структуры белков (например, ферментов или компонентов мембран, энергетического обеспечения реакций метаболизма и жизненно важных процессов, искажающих дифференцировку и функции клеток, тканей и органов. Нарушения морфогенетических процессов (пролиферация, миграция, дифференцировка и гибель клеток) приводят к аплазии (отсутствие органа при наличии его сосудистой ножки) или гипоплазии (недоразвитию) органа или его части, задержке слияния эмбриональных структур (например, расщелины нёба, губы; спинномозговые и черепно-мозговые грыжи), персистированию эмбриональных структур, к атрезии и гетеротопии (наличие клеток и/или тканей в другом органе или в тех зонах органа, где их в норме не должно быть [например, участки ткани поджелудочной железы в дивертикуле Меккеля]) и т.д. и т.п. (см., например, подраздел «Категории ВПР»). КАТЕГОРИИ ВПР Наиболее частые категории ВПР представлены на рис. 3–16. 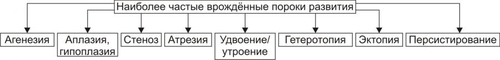 Рис. 3–16. Наиболее частые категории врождённых пороков развития. Агенезия — полное отсутствие органа (например, тимуса, почки, глаз). Аплазия и гипоплазия — отсутствие или значительное уменьшение органа при наличии его сосудистой ножки и нервов (например, одной почки, селезёнки, лёгкого, конечности, кишечника). Атрезия — полное отсутствие канала или естественного отверстия (например, атрезия наружного слухового прохода, пищевода, ануса). Гетеротопия — перемещение клеток, тканей или части органа в другую ткань или орган (например, клеток поджелудочной железы в дивертикул Меккеля, хромаффинных клеток в ткань лёгких). Персистирование — сохранение эмбриональных структур, исчезающих в норме к определённому этапу развития (например, открытый артериальный проток у годовалого ребёнка, крипторхизм) Стеноз — сужение просвета отверстия или канала (например, клапанного отверстия сердца, привратника желудка, фрагмента кишечника). Удвоение (утроение) — увеличение числа органов или его части (например, удвоение матки, мочеточников). Эктопия — необычное расположение органа (например, почки в малом тазу, сердца — вне грудной клетки). МЕТОДЫ ДИАГНОСТИКИ Методы диагностики наследственных форм патологии и методы изучения их патогенеза приведены на рис. 3–17. 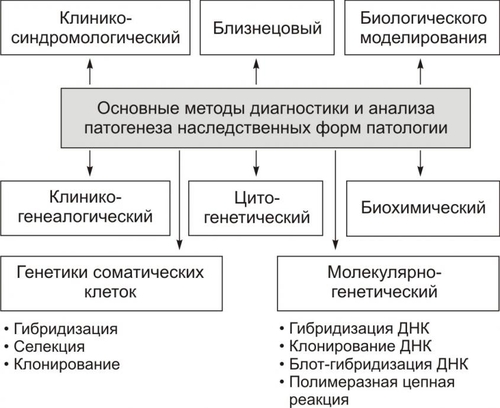 Рис. 3–17. Методы диагностики и анализа наследственных форм патологии. Ниже коротко перечислены цели и возможности клинико–синдромологического и клиникогенеалогического методов (в том числе составление родословной и изучение близнецов), а также методов цитогенетической, биохимической и молекулярной диагностики. КЛИНИКОСИНДРОМОЛОГИЧЕСКИЙ МЕТОД Клиникосиндромологический метод позволяет выявлять морфологические, биохимические и функциональные признаки наследственных форм патологии (например, дефицит плазменного фактора VIII при подозрении на гемофилию A; кариотип 45,Х0 при подозрении на синдром Шерешевского–Тёрнера; поражения скелета, ССС и глаз при подозрении на синдром Марфана). КЛИНИКОГЕНЕАЛОГИЧЕСКИЙ МЕТОД Клиникогенеалогический метод выявляет патологические признаки и прослеживает особенности их передачи в поколениях при составлении родословной. Цель — установление закономерностей наследования признака: • определение типа наследования (доминантного, рецессивного, ауто или гоносомного); • выявление носителей гена, определяющего развитие исследуемого признака (болезни); • оценка пенетрантности (частоты проявления) гена — «носителя» патогенного признака; • определение генетического риска (вероятности рождения больного ребёнка). СОСТАВЛЕНИЕ РОДОСЛОВНОЙ Составление родословной начинают со сбора сведений о семье консультирующегося или пробанда. Консультирующимся называется лицо, обратившееся к врачу или первое попавшееся в поле зрения исследователя лицо. Пробанд — больной или носитель изучаемого признака. Во многих случаях консультирующийся и пробанд являются одним и тем же лицом. Дети одной родительской пары называются сибсами (братья и сёстры). Семьёй в узком смысле называют родительскую пару и их детей, но иногда и более широкий круг кровных родственников, хотя в последнем случае лучше применять термин род. Примеры родословных при разных типах наследования представлены на рис. 3–8—3–12. БЛИЗНЕЦОВЫЙ МЕТОД Близнецовый метод базируется на сравнительном анализе частоты определённого признака в разных группах близнецов, а также в сопоставлении с партнёрами монозиготных пар между собой и общей популяцией. Идентичность близнецов по анализируемому признаку обозначают как конкордантность, а отличие — как дискордантность. Роль наследственности и факторов среды в возникновении патологии у близнецов оценивают по специальным формулам. ЦИТОГЕНЕТИЧЕСКАЯ ДИАГНОСТИКА Цитогенетическая диагностика основана на микроскопическом изучении хромосом с целью выявления структурных нарушений в хромосомном наборе (кариотипирование). В качестве материала используют тканевые культуры с большим числом делящихся клеток, чаще лимфоциты периферической крови. Хромосомы на стадии метафазы изучают при помощи специальных методов окрашивания и составляют идиограммы (систематизированные кариотипы с расположением хромосом от наибольшей к наименьшей), что позволяет выявлять геномные и хромосомные мутации. БИОХИМИЧЕСКАЯ ДИАГНОСТИКА Биохимическая диагностика базируется на изучении биохимических показателей, отражающих сущность болезни (например, активность ферментов, наличие патологических метаболитов, концентрация компонентов ферментативной реакции). Объекты исследования: • метаболиты в биологических жидкостях и клетках (например, фенилаланин при фенилпировиноградной олигофрении; кетоновые тела (КТ) при сахарном диабете). • аномальные белки (например, Hb при гемоглобинопатиях). • дефектные ферменты (например, холинэстераза, глутатион пероксидаза, каталаза). Этапы исследования: • первый — использование скрининговых программ диагностики (например, тонкослойной хроматографии, электрофореза, микробиологических методов); • второй — применение подтверждающих методов (например, флюорометрических, спектрофотометрических, количественного определения метаболитов, тестирования активности фермента). МОЛЕКУЛЯРНАЯ ДИАГНОСТИКА При помощи методов ДНКдиагностики устанавливают последовательность расположения отдельных нуклеотидов, выделяют гены и их фрагменты, устанавливают их наличие в изучаемых клетках. К числу наиболее эффективных методов относятся гибридизация ДНК (блоттинг, in situ и т.д.), клонирование ДНК, полимеразная цепная реакция (ПЦР). ГИБРИДИЗАЦИЯ ДНК Для определения порядка расположения нуклеотидов в исследуемом генетическом материале изучаемую ДНК помещают в специальную среду, где происходит контакт ДНК с нитями другой нуклеиновой кислоты. В случае комплементарности какихлибо двух нитей происходит их «noeaea». При специальных исследованиях используют генетические «ciiau» — фрагменты меченной радиоактивным изотопом однонитевой ДНК с известной последовательностью нуклеотидов. БЛОТГИБРИДИЗАЦИЯ Для выявления интересующих (в том числе мутантных) генов ДНК подвергают рестрикции. Полученные фрагменты ДНК подразделяют по молекулярной массе, денатурируют и переносят на носитель (нейлоновую или иную мембрану). Фиксированную на носителе в виде пятна ДНК гибридизируют с меченым радиоактивным изотопом ДНК или РНКзондом. В результате определяют положение аномального фрагмента ДНК. КЛОНИРОВАНИЕ ДНК С помощью специализированных ферментов (ДНКрестриктаз) подразделяют нить ДНК на отдельные группы генов или на единичные гены. Для изучения признаков (в том числе патологических), кодируемых данными генами, особенностей транскрипции и трансляции создают нужное количество копий данного гена. ПОЛИМЕРАЗНАЯ ЦЕПНАЯ РЕАКЦИЯ ПЦР (специфическая амплификация небольшого участка ДНК) применяют для изучения регионов предполагаемых мутаций и других особенностей структуры ДНК. Для исследования можно использовать любой биологический материал, содержащий ДНК (например, кусочек ткани, капля или пятно крови, смыв полости рта, луковица корня волос). На первом этапе исследуемую ДНК расщепляют на две нити при нагревании до 95–98 °C. Затем одну из нитей гибридизируют и стимулируют синтез последовательности, комплементарной исследуемой ДНК (с помощью термофильной ДНКполимеразы). Циклы реакции повторяют до накопления нужного количества ДНК. Эту методику разработал и предложил Кэри Муллис. БИОЛОГИЧЕСКОЕ МОДЕЛИРОВАНИЕ Биологическое моделирование проводят для анализа возможных генетических дефектов человека с использованием в качестве объекта исследования животных (здоровых или мутантных), а также для изучения возможных мутагенных и тератогенных эффектов ЛС и других агентов, для разработки методов генной инженерии. ПРИНЦИПЫ ЛЕЧЕНИЯ При лечении наследственных болезней (при соблюдении индивидуального характера помощи) применяют три основных подхода: этиотропный, патогенетический и симптоматический. ЭТИОТРОПНАЯ ТЕРАПИЯ Этиотропный подход направлен на устранение причины заболевания. С этой целью разрабатываются, апробируются и частично могут быть применены методы коррекции генетических дефектов, называемые генной терапией. Генная терапия бурно развивается в последние годы, что является непосредственным следствием успешно выполняемой международной программы «Геном Человека». В общем виде целью генной терапии является внесение в клеточный геном поражённых органов нормально экспрессируемого «здорового» гена, восполняющего функцию мутантного («больного») гена. Непременным условием генной терапии конкретной наследственной патологии является наличие клонированного (и нормального!) гена, ответственного за развитие заболевания (моногенного, как правило). Конечная задача — внедрение нормального гена в геном клеток поражённого органа. Эта процедура выполняется при помощи трансфекции — введения в геном клетки вектора, содержащего нужный и здоровый ген человека. В качестве векторов обычно применяют модифицированные (дефектные по репликации) вирусы (ретро–, адено– и др.). Процедуру трансфекции обычно выполняют ex vivo: при культивировании клеток больного человека; при успешном внедрении вектора с нужным геном в геном культивируемых клеток последние вводят тем или иным способом в нужный орган пациента. В качестве клетокмишеней для генной терапии применяют только соматические (но не половые) клетки — носители патогенных генов. В настоящее время апробированы и утверждены сотни генотерапевтических протоколов для лечения моногенных и многофакторных заболеваний (например, тяжёлого комбинированного иммунодефицита, семейной гиперхолестеринемии, муковисцидоза, некоторых мышечных дистрофий, различных онкологических и инфекционных заболеваний). ПАТОГЕНЕТИЧЕСКАЯ ТЕРАПИЯ Цель патогенетической терапии — разрыв звеньев патогенеза. Для достижения этой цели применяют несколько методов. • Заместительная терапия — введение в организм дефицитного вещества (не синтезирующегося в связи с аномалией гена, который контролирует продукцию данного вещества; например, инсулина при СД, соответствующих ферментов при гликогенозах и агликогенозах, антигемофильного глобулина человека при гемофилии). • Коррекция метаболизма путём: † ограничения попадания в организм веществ, метаболически не усваивающихся (например, фенилаланина или лактозы); † выведения из организма метаболитов, накапливающихся в нём в избытке (например, фенилпировиноградной кислоты или холестерина); † регуляции активности ферментов (например, подавление активности креатинфосфокиназы [КФК] при отдельных видах миодистрофий, активация липопротеин липазы [ЛПЛаза] крови при гиперхолестеринемии). • Хирургическая коррекция дефектов (например, создание шунта между нижней полой и воротной венами у пациентов с «гепатотропными» гликогенозами). СИМПТОМАТИЧЕСКАЯ ТЕРАПИЯ Симптоматическая терапия направлена на устранение симптомов, усугубляющих состояние пациента (например, применение веществ, снижающих вязкость секретов экзокринных желёз при муковисцидозе; хирургическое удаление дополнительных пальцев и/или перемычек кожи между ними при поли– и синдактилии; выполнение пластических операций при дефектах лица, пороках сердца и крупных сосудов). ПРОФИЛАКТИКА Цель профилактического направления медицинской генетики — предотвратить или снизить риск возникновения заболеваний: • вновь появившихся вследствие мутации в половых клетках родителей; • унаследованных от предыдущих поколений; • возникающих в связи с наследственной предрасположенностью под влиянием факторов среды. Эта цель может быть достигнута на четырёх этапах индивидуального развития: • прегаметическом (охрана здоровья человека в репродуктивном возрасте, охрана окружающей среды); • презиготном (например, искусственная инсеминация, медикогенетическое консультирование); • пренатальном (все виды дородовой диагностики); • постнатальном (раннее выявление, «профилактическое лечение» — до развития симптомов заболевания; охранительный режим). МЕТОДЫ ПРОФИЛАКТИКИ МЕДИКОГЕНЕТИЧЕСКОЕ КОНСУЛЬТИРОВАНИЕ Медикогенетическое консультирование является основным видом профилактики врождённой и наследственной патологии. Задача консультирования: сформулировать прогноз для потомства, течения заболевания, качества жизни и здоровья. ПРЕНАТАЛЬНАЯ ДИАГНОСТИКА Пренатальная диагностика осуществляется в I и II триместрах беременности (в периоды, когда возможно прерывание беременности при обнаружении патологии плода). Методы диагностики • Ультразвуковое исследование (УЗИ) позволяет установить наличие беременности, количество плодов, выявлять грубые аномалии плода. • Биохимическое исследование сыворотки крови матери: определение концентрации • Фетоскопия обеспечивает прямое наблюдение плода с помощью специальной оптической системы, позволяет диагностировать заболевания кожи, нарушения развития половых органов, дефекты лица, конечностей и пальцев, производить биопсию тканей плода. • Цитогенетические, биохимические и молекулярногенетические методы применяют на материале, полученном при следующих вмешательствах: † Амниоцентез позволяет получить амниотическую жидкость путём пункции амниона, проводят на 15–17-й неделях беременности. † Кордоцентез обеспечивает возможность взятия крови плода путём пункции пуповины, проводят на 15–17-й неделе беременности. † Хорионбиопсия — взятие для исследования ворсин хориона, клетки которого генетически идентичны клеткам развивающегося организма, проводят на 6–11-й неделе беременности. ПРЕКЛИНИЧЕСКАЯ ДИАГНОСТИКА Преклиническая диагностика — скрининг с целью ранней диагностика наследственных болезней обмена веществ у новорождённых. Применяют относительно простые, но надёжные методы. Скринингу подлежат наследственные заболевания обмена: приводящие к гибели или инвалидизации (без раннего выявления и своевременного лечения); встречающиеся с частотой не реже чем 1:20 000–1:50 000. новорождённых; имеющие адекватные и экономичные методы предварительного выявления; подвергающиеся эффективному лечению, реабилитации и адаптации. ДИСПАНСЕРИЗАЦИЯ Диспансеризация семей с наследственной патологией проводится с целью предупреждения рождения больного ребёнка или зачатия аномального плода (первичная профилактика). КОНТРОЛЬ МУТАГЕННОЙ ОПАСНОСТИ Контроль мутагенной опасности факторов окружающей среды реализуется путём предотвращения образования, снижения содержания, длительности и/или силы действия на организм химических, физических и биологических мутагенных агентов. Достигается комплексом организационных и гигиенических мер на производстве, в учреждениях и быту (например, возведением очистных сооружений; применением спецодежды, очисткой воздуха, воды и продуктов питания; использованием средств противорадиационной защиты).
|