Лекции по биологии (1 семестр). Лекция 1 Типы клеточной организации
 Скачать 1.41 Mb. Скачать 1.41 Mb.
|

|
Генные мутации. Понятие о генных болезнях.
Изменчивость – есть общее свойство живых организмов, заключающееся в изменении наследственных признаков в ходе онтогенеза (индивидуального развития). Изменчивость организмов делят на два крупных типа: 1. фенотипическую, не затрагивающую генотип и не передающуюся по наследству; 2. генотипическую, изменяющую генотип и поэтому передающуюся по наследству. Генотипическая изменчивость подразделяется на комбинативную и мутационную. Мутационная изменчивость включает геномные, хромосомные и генные мутации. Геномные мутации подразделяется на полиплоидию и анеуплоидию Хромосомные мутации подразделяется на делеции, дупликации, инверсии, транслокации
Фенотипическая изменчивость (или ненаследственная, модификационная) – это изменение фенотипических признаков организма под действием факторов внешней среды, без изменения генотипа. Например: окраска шерсти у гималайского кролика в зависимости от температуры среды обитания. Норма реакции – это диапазон изменчивости, в пределах которого один и тот же генотип способен давать различные фенотипы.
Адаптивный характер модификаций заключается в том, что модификационная изменчивость позволяет организму адаптироваться к изменяющимся условиям среды. Поэтому модификации всегда полезны. Если во время эмбриогенеза на организм воздействуют неблагоприятные факторы, то могут появляться фенотипические изменения, выходящие за пределы нормы реакции и не носящие адаптивного характера, их называют морфозы развития. Например, ребёнок рождается без конечностей или с заячьей губой. Фенокопии – это морфозы развития, которые очень трудно отличить от наследственных изменений (заболеваний). Например: если беременная женщина переболела краснухой, у неё может родиться ребёнок с катарактой. Но эта патология может появиться и в результате мутации. В первом случае речь идет о фенокопии. Диагноз «фенокопия» важен для будущего прогноза, так как при фенокопии генетический материал не изменяется, то есть остается в норме.
Комбинативная изменчивость – это возникновение у потомков новых комбинаций генов, которых не было у их родителей. Комбинативная изменчивость связана:
Значение комбинативной изменчивости – обеспечивает генетическое разнообразие особей в пределах вида, что важно для естественного отбора и эволюции.
Гюго де Фриз голландский ученый ввел в 1901 году термин "мутация". Мутация – это явление прерывистого скачкообразного изменения наследственного признака. Процесс возникновения мутаций называется мутагенез, а организм, который приобретает новые признаки в процессе мутагенеза, называется – мутант. Основные положения теории мутаций по Гюго де Фризу.
Генные мутации или точковые мутации – это мутации, которые возникают в генах на уровне нуклеотидов, при этом изменяется структура гена, изменяется молекула мРНК, изменяется последовательность аминокислот в белке, в организме изменяется признак. Виды генных мутаций:
Пример: замена глутаминовой кислоты на валин в молекуле гемоглобина. ЦТТ – глутаминовая кислота, ЦАТ – валин Если такая мутация происходит в гене, который кодирует β цепь белка гемоглобина, то в β цепь вместо глютаминовой кислоты включается валин → в результате такой мутации изменяются свойства и функции белка гемоглобина и вместо нормального HbA появляется HbS, в результате у человека развивается серповидноклеточная анемия (форма эритроцитов изменяется).
Если в результате генной мутации у организма будет появляться новый признак (например, полидактилия), то они называются неоморфные. если в результате генной мутации организм утрачивает признак (например, при ФКУ исчезает фермент) то они называются аморфные.
Механизмы возникновения генных мутаций (замена, вставка, выпадение). ДНК состоит из 2-х полинуклеотидных цепей. Сначала изменение возникает в 1-й цепи ДНК – это полумутационное состояние или “первичное повреждение ДНК”. Каждую секунду в клетке имеет место 1 первичное повреждение ДНК. Когда повреждение переходит на вторую цепь ДНК то, говорят о том, что произошла фиксация мутации, то есть возникла “полная мутация”. Первичные повреждения ДНК возникают при нарушении механизмов репликации, транскрипции, кроссинговера
У человека частота мутаций = 1х10–4 – 1х10–7, то есть в среднем 20–30% гамет у человека в каждом поколении являются мутантными. У дрозофилы частота мутаций = 1х10–5, то есть 1 гамета из 100 тысяч несет генную мутацию.
Генные мутации встречаются у всех организмов, гены мутируют в различных направлениях, а также с различной частотой. Гены, которые редко мутируют называются – стабильные, а гены, которые часто мутируют называются – мутабельные.
Мутирование происходит в самых различных направлениях, т.е. случайно. Однако эти случайности подчиняются закономерности, обнаруженной в 1920г. Вавиловым. Он сформулировал закон гомологичных рядов в наследственной изменчивости. "Виды и роды генетически близкие характеризуются сходными рядами наследственной изменчивости с такой правильностью, что, зная ряд форм в пределах одного вида, можно предвидеть существование параллельных форм у других видов и родов". Этот закон позволяет предсказать наличие определённого признака у особей различных родов одного семейства. Так было предсказано наличие в природе безалкалоидного люпина, т.к. в семействе бобовых есть роды бобов, гороха, фасоли, не содержащие алкалоиды. В медицине закон Вавилова позволяет использовать животных, генетически близких человеку, в качестве генетических моделей. На них ставят эксперименты по изучению генетических болезней. Например, катаракта изучается на мышах и собаках; гемофилия – на собаках, врождённая глухота – на мышах, морских свинках, собаках. Закон Вавилова позволяет предвидеть появление индуцированных мутаций, неизвестных науке, которые могут использоваться в селекции для создания ценных для человека форм растений.
Репарация ДНК – удаление первичных повреждений из ДНК и замена их нормальными структурами. Выделяют две формы репарации: световую и темновую А. Световая репарация (или ферментативная фотореактивация). Ферменты репарации активны только в присутствии света. Эта форма репарации направлена на удаление первичных повреждений ДНК вызванных действием УФЛ. П 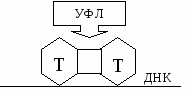 од действием УФЛ в ДНК активируются пиримидиновые азотистые основания, что приводит к тому, что возникают связи между пиримидиновыми азотистыми основаниями, которые располагаются рядом в одной цепи ДНК, то есть образуются пиримидиновыедимеры. Чаще всего возникают связи: Т=Т; Т=Ц; Ц=Ц. од действием УФЛ в ДНК активируются пиримидиновые азотистые основания, что приводит к тому, что возникают связи между пиримидиновыми азотистыми основаниями, которые располагаются рядом в одной цепи ДНК, то есть образуются пиримидиновыедимеры. Чаще всего возникают связи: Т=Т; Т=Ц; Ц=Ц.В норме в ДНК пиримидиновых димеров нет. Образование их приводит к тому, что искажается наследственная информация и нарушается нормальный ход репликации и транскрипции, что приводит впоследствии к генным мутациям. Суть фотореактивации: в ядре существуют специальный (фотореактивирующий) фермент, который активен только в присутствии света, этот фермент разрушает пиримидиновые димеры, то есть разрывает связи, которые возникли между пиримидиновыми азотистыми основаниями под действием УФЛ. Темновая репарация происходит в темноте и на свету, то есть активность ферментов не зависит от присутствия света. Она делится на дорепликативная репарацию и пострепликативную репарацию. Дорепликативная репарация происходит до репликации ДНК, в этом процессе участвует много ферментов:
Допустим, в ДНК имеется первичное повреждение. 1  этап. Фермент эндонуклеаза находит поврежденный участок и разрезает его. этап. Фермент эндонуклеаза находит поврежденный участок и разрезает его. 2  этап. Фермент экзонуклеаза удаляет поврежденный участок из ДНК (эксцизия) в результате образуется брешь. этап. Фермент экзонуклеаза удаляет поврежденный участок из ДНК (эксцизия) в результате образуется брешь. 3 этап. Фермент ДНК полимераза синтезирует недостающий участок. Синтез происходит по принципу комплементарности. 4  этап. Ферменты лигазы соединяют или сшивают вновь синтезированный участок с цепью ДНК. Таким образом, первичное повреждение в ДНК устраняется. этап. Ферменты лигазы соединяют или сшивают вновь синтезированный участок с цепью ДНК. Таким образом, первичное повреждение в ДНК устраняется. Пострепликативная репарация. Допустим, в ДНК имеется первичное повреждение. 1  этап. Начинается процесс репликации ДНК. Фермент ДНК-полимераза синтезирует новую цепь полностью комплементарную старой неповрежденной цепи. этап. Начинается процесс репликации ДНК. Фермент ДНК-полимераза синтезирует новую цепь полностью комплементарную старой неповрежденной цепи. 2  этап. Фермент ДНК полимераза синтезирует другую новую цепь, но участок, где находится повреждение, он обходит. В результате во второй новой цепи ДНК образовалась брешь. этап. Фермент ДНК полимераза синтезирует другую новую цепь, но участок, где находится повреждение, он обходит. В результате во второй новой цепи ДНК образовалась брешь. 3  этап. По окончании репликации фермент ДНК полимераза синтезирует недостающий участок комплементарно новой цепи ДНК. этап. По окончании репликации фермент ДНК полимераза синтезирует недостающий участок комплементарно новой цепи ДНК. 4   этап. Затем фермент лигаза соединяют вновь синтезированный участок с цепью ДНК, где имелась брешь. Таким образом, первичное повреждение ДНК не перешло на другую новую цепь, то есть не произошла фиксация мутации. этап. Затем фермент лигаза соединяют вновь синтезированный участок с цепью ДНК, где имелась брешь. Таким образом, первичное повреждение ДНК не перешло на другую новую цепь, то есть не произошла фиксация мутации.В дальнейшем первичное повреждение ДНК может быть ликвидировано в ходе дорепликативной репарации.
Способность к репарации у организмов выработалась и закрепилась в ходе эволюции. Чем выше активность репарирующих ферментов, тем стабильнее наследственный материал. За ферменты репарации отвечают соответствующие гены, поэтому если происходит мутация в этих генах, то снижается активность репарирующих ферментов. У человека при этом возникают тяжелые наследственные заболевания, которые связаны со снижением активности репарирующих ферментов. Таких заболеваний у человека больше 100. Некоторые из них: Анемия Фанкони – уменьшение количества эритроцитов, потеря слуха, нарушения в ССС, деформация пальцев, микроцефалия. Сидром Блума – малый вес новорождённого, замедление роста, повышенная восприимчивость в вирусной инфекции, повышенный риск онкологических заболеваний. Характерный признак: при непродолжительном пребывании на солнечном свету на коже лица появляется пигментация в форме бабочки (расширение кровеносных капилляров). Пигментная ксеродермия – на коже от света появляются ожоги, которые скоро перерождаются в рак кожи (у таких больных рак возникает в 20.000 раз чаще). Больные вынуждены жить при искусственном освещении. Частота заболевания – 1 : 250.000 (Европа, США), и 1 : 40.000 (Япония) Два вида прогерий – преждевременное старение организма.
Генные болезни (или молекулярные болезни) достаточно широко представлены у человека, их насчитывается более 1000. Особую группу среди них составляют врожденные дефекты обмена веществ. Впервые эти заболевания описал А. Гарод в 1902 году. Симптоматика этих заболеваний различна, но всегда имеет место нарушение превращения веществ в организме. При этом одни вещества будут в избытке, другие в недостатке. Например, в организм поступает вещество (А) и превращается далее под действием ферментов в вещество (В). Далее вещество (В) должно превращаться в вещество (С), но этому мешает мутационный блок (  ), в результате вещество (С) будет в недостатке, а вещество (В) в избытке. ), в результате вещество (С) будет в недостатке, а вещество (В) в избытке. А → В С Примеры некоторых болезней, обусловленных врожденным дефектом обмена веществ. ФКУ (фенилкетонурия, врожденное слабоумие). Генное заболевание, наследуется по аутосомно-рецессивному типу, встречается с частотой = 1:10.000. Фенилаланин является незаменимой аминокислотой для построения белковой молекулы и, кроме того, служит предшественником гормонов щитовидной железы (тироксина), адреналина и меланина. Аминокислота фенилаланин в клетках печени должна превращаться с помощью фермента (фенилаланин-4-гидроксилазы) в тирозин. Если отсутствует фермент, отвечающий за данное превращение, или снижена его активность то содержание фенилаланина в крови будет резко повышено, а содержание тирозина понижено. Избыток фенилаланина в крови приводит к появлению его производных (фенилуксусной, фенилмолочной, фенилпировиноградной и других кетоновых кислот), которые выделяются с мочой, а также оказывают токсическое воздействие на клетки центральной нервной системы, что приводит к слабоумию. При своевременной постановке диагноза и переводе младенца на диету, лишенную фенилаланина, развитие заболевания можно предупредить. Альбинизм общий. Генное заболевание, наследуется по аутосомно-рецессивному типу. В норме аминокислота тирозин участвует в синтезе тканевых пигментов. Если возникает мутационный блок, отсутствует фермент или снижена его активность, то тканевые пигменты не синтезируются. В этих случаях кожа имеет молочно-белый цвет, волосы очень светлые, вследствие отсутствия пигмента в сетчатке просвечивают кровеносные сосуды, глаза имеют красновато-розовый цвет, и повышенную чувствительность к свету. Алькапнонурия. Генное заболевание, наследуется по аутосомно-рецессивному типу, встречается с частотой = 3-5:1.000.000. Заболевание связано с нарушением превращения гомогентизиновой кислоты, в результате чего эта кислота накапливается в организме. Выделяясь с мочой, эта кислота приводит к развитию заболеваний почек, кроме того, подщелоченная моча при этой аномалии быстро темнеет. Также заболевание проявляется окрашиванием хрящевых тканей, в пожилом возрасте развивается артрит. Таким образом, заболевание сопровождается поражением почек и суставов. Генные болезни, связанные с нарушением обмена углеводов. Галактоземия. Генное заболевание, наследуется по аутосомно-рецессивному типу, встречается с частотой = 1:35.000-40.000 детей. В крови новорождённого содержится моносахарид галактоза, который образуется при расщеплении дисахарида молока лактозы на глюкозу и галактозу. Галактоза непосредственно не усваивается организмом, она должна быть переведена специальным ферментом в усваиваемую форму – глюкоза-1-фосфат. Наследственная болезнь галактоземия обусловлена нарушением функции гена, контролирующего синтез белка-фермента, превращающего галактозу в усваиваемую форму. В крови больных детей будет очень мало этого фермента и много галактозы, что устанавливается биохимическим анализом. Если диагноз поставлен в первые дни после рождения ребенка, то его кормят смесями, где нет молочного сахара, и ребёнок нормально развивается. В противном случае ребёнок вырастает слабоумным. Муковисцидоз. Генное заболевание, наследуется по аутосомно-рецессивному типу, встречается с частотой = 1:2.000-2.500. Заболевание связано с мутацией гена, который отвечает за белок-переносчик, встроенный в плазматическую мембрану клеток. Этот белок регулирует проницаемость мембраны к ионам Na и Ca. Если нарушена проницаемость этих ионов в клетках экзокринных желез, то железы начинают вырабатывать густой, вязкий секрет, который закрывает протоки экзокринных желез. Выделяют легочную и кишечную формы муковисцидоза. Синдром Марфана. Генное заболевание, наследуется по аутосомно-доминантному типу. Связано с нарушением обмена белка фибриллина в соединительной ткани, что проявляется комплексом признаков: «паучьи» пальцы (арахнодактилия), высокий рост, подвывих хрусталика, пороки сердца и сосудов, повышенный выброс в кровь адреналина, сутулость, впалая грудь, высокий свод стопы, слабость связок и сухожилий и т.д. Впервые описано в 1896 году французским педиатром Антонио Марфаном. ЛЕКЦИЯ 10 Структурные мутации хромосом. 1. Структурные мутации хромосом (хромосомные аберрации). Выделяют следующие виды хромосомных аберраций. – делеции – дупликации – инверсии – кольцевые хромосомы – транслокации – транспозиции При данных мутациях изменяется структура хромосом, изменяется порядок расположения генов в хромосомах, изменяется доза генов в генотипе. Эти мутации встречаются у всех организмов, они бывают:
Если мутация затрагивает две хромосомы, говорят о межхромосомных перестройках. Если мутация затрагивает 1 хромосому, говорят о внутрихромосомных перестройках.
Делеция (нехватка) – потеря участка хромосомы.
В гомозиготном состоянии делеции всегда летальны, в гетерозиготном состоянии они проявляются множественными пороками развития. Выявление делеций:
Впервые делеция была изучена у мушки дрозофилы, при этом произошла потеря участка Х хромосомы. В гомозиготном состоянии эта мутация летальна, а в гетерозиготном состоянии она проявляется фенотипически вырезкой на крыле (Notch-мутация). При анализе этой мутации было выявлено особое явление, которое получило название псевдодоминирование. При этом фенотипически проявляется рецессивный аллель, так как участок хромосомы с доминантным аллелем утрачен вследствие делеции. У человека делеции чаще происходят в хромосомах с 1 по 18. Например, делеция короткого плеча пятой хромосомы в гетерозиготном состоянии проявляется фенотипически, как синдром "кошачьего крика". Ребёнок рождается с большим числом патологий, живет от 5 дней до месяца (очень редко до 10 лет), его плач напоминает резкое мяуканье кота. В 21 или 22 хромосоме стволовых кроветворных клеток может произойти интерстициальная делеция. В гетерозиготном состоянии она проявляется фенотипически как злокачественная анемия.
Дупликация – удвоение какого-то участка хромосомы (этот участок может повторяться многократно). Дупликации могут быть прямыми и обратными. При данных мутациях увеличивается доза генов в генотипе, и в гомозиготном состоянии эти мутации летальны. В гетерозиготном состоянии они проявляются множественными пороками развития. Однако эти мутации могли играть определенную роль в ходе эволюции. Таким образом могли возникнуть семейства генов гемоглобина. Возможно, многократно повторяющиеся последовательности нуклеотидов ДНК появились в результате дупликаций. Выявление дупликаций:
Инверсия – отрыв участка хромосомы, поворот его на 180° и присоединение на старое место. При инверсиях доза генов не меняется, но изменяется порядок расположения генов в хромосоме, т.е. изменяется группа сцепления. Концевых инверсий не бывает. В гомозиготном состоянии инверсии летальны, в гетерозиготном состоянии они проявляются множественными пороками развития. Выявление инверсий:
Инверсии бывают 2 видов:
При перицентрической инверсии может изменяться конфигурация хромосомы (если концы поворачиваемых участков не симметричны). А это делает невозможным в последующем конъюгацию. Фенотипическое проявление инверсий наиболее мягкое по сравнению с другими хромосомными абберациями. Если рецессивные гомозиготы погибают, то у гетерозигот чаще всего наблюдается бесплодие. Кольцевые хромосомы. В норме в кариотипе человека кольцевых хромосом нет. Они могут появляться при действии на организм мутагенных факторов, особенно радиоактивного облучения. При этом в хромосоме происходит 2 разрыва, и образовавшийся участок замыкается в кольцо. Если кольцевая хромосома содержит центромеру, то образуется – центрическое кольцо. Если центромеры нет, то образуется – ацентрическое кольцо, оно разрушается ферментами и не наследуется. Выявляются кольцевые хромосомы при кариотипировании. В гомозиготном состоянии эти мутации летальны, а в гетерозиготном состоянии фенотипически проявляются, как делеции. Кольцевые хромосомы являются маркерами радиоактивного облучения. Чем больше доза радиоактивного облучения, тем больше кольцевых хромосом, и тем хуже прогноз.
Транслокация – это перемещение участка хромосомы. Бывают взаимные (реципрокные) и не взаимные (транспозиции) транслокации. Реципрокные транслокации происходят в тех случаях, когда две негомологичные хромосомы обмениваются своими участками. Особую группу транслокаций составляют робертсоновские транслокации (центрические слияния). Им подвергаются акроцентрические хромосомы – они теряют короткие плечи, а их длинные плечи соединяются. 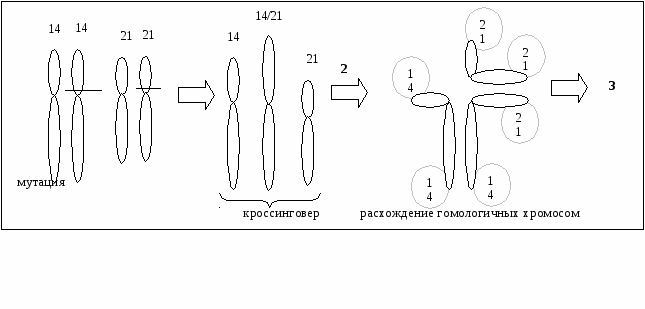 Причина 4-5% случаев рождения ребёнка-дауника – робертсоновские транслокации. При этом происходит перемещение длинного плеча 21 хромосомы на одну из хромосом группы D (13, 14, 15, чаще вовлекается 14 хромосома). Типы яйцеклеток сперматозоид зигота Последствия 14 + 14, 21 14,14,21 моносомия 21 (леталь) 14/21,21 + 14, 21 14/21,21,14,21 трисомия 21 (дауник) 21 + 14, 21 21,14,21, моносомия 14 (леталь) 14,14/21 + 14, 21 14,14/21,14,21 трисомия 14 (леталь) 14/21 + 14, 21 14/21,14,21 фенотипически здоров Как видим, женщина с робертсоновской транслокацией может родить здорового ребенка. Потеря коротких плеч не влияет ни на что, так как там находятся ядрышкообразующие зоны, а они есть и в других хромосомах. У больного с транслокационной формой синдрома Дауна в клетках 46 хромосом. В яичнике после транслокации будет 45 хромосом. Однако при сбалансированной мутации у женщины будет 45 хромосом. Выявление транслокаций:
Если транслокации не носят характера взаимности, то говорят о транспозиции. Особую группу транспозонов составляют Мобильные Генетические Элементы (МГЭ), или прыгающие гены, которые обнаружены у всех организмов. У мушки дрозофилы они составляют 5% генома. У человека МГЭ объединяют в семейство ALU. МГЭ состоят из 300- 400 нуклеотидов, повторяющихся в геноме у человека 300 тыс. раз. На МГЭ концах находятся повторы нуклеотидов, состоящие при из 50-100 нуклеотидов. Повторы могут быть прямыми и обратными. Повторы нуклеотидов, по-видимому, влияют на перемещение МГЭ. Выделяют два варианта перемещения МГЭ по геному. 1. с помощью процесса обратной транскрипции. Для этого необходим фермент обратная транскриптаза (ревертаза). Этот вариант протекает в несколько этапов:
2. с помощью фермента транспозазы, который вырезает МГЭ и переносит его в другую хромосому или в другое место этой же хромосомы В ходе эволюции МГЭ играли положительную роль, т.к. они осуществляли перенос генетической информации от одних видов организмов к другим. Важную роль в этом играли ретровирусы, которые содержат в качестве наследственного материала РНК, а также содержат обратную транскриптазу. МГЭ перемещаются по геному очень редко, одно перемещение на сотни тысяч событий в клетке (частота перемещений 1 х 10–5). В каждом конкретном организме МГЭ положительной роли не играют, т.к. перемещаясь по геному, они изменяют работу генов, вызывают генные и хромосомные мутации.
Индуцированные мутации возникают при действии на организм мутагенных факторов, которые делятся на 3 группы:
Так ионизирующее излучение может действовать непосредственно на молекулы ДНК и РНК, вызывая в них повреждения (генные мутации). Косвенное воздействие этого мутагена на наследственный аппарат клеток заключается в образовании генотоксических веществ (Н2О2, ОН-, О2-,).
Радиационные мутации это мутации, вызванные радиацией. В 1927 году американский генетик, Генрих Мелёр впервые показал, что облучение рентгеновскими лучами приводит к существенному увеличению частоты мутаций у дрозофилы. Эта работа положила начало новому направлению в биологии – радиационной генетике. Благодаря многочисленным работам, проведенным за последние десятилетия, мы теперь знаем, что при попадании элементарных частиц (кванты, электроны, протоны и нейтроны) в ядро происходит ионизация молекул воды с образованием свободных радикалов (ОН-, О2-). Обладая большой химической активностью, они вызывают разрывы ДНК, повреждение нуклеотидов или их разрушение; всё это приводит к возникновению мутаций. Так как человек является открытой системой, то различные факторы загрязнения окружающей среды могут попадать в человеческий организм. Многие из этих факторов могут изменять или повреждать наследственный материал живых клеток. Последствия воздействия этих факторов столь серьезны, что человечество не может игнорировать загрязнение окружающей среды.
Впервые мутационную теорию рака в 1901 году предложил Гюго Де Фриз. В наши дни существует много теорий канцерогенеза. Одна из них генная теория канцерогенеза. Известно, что в геноме человека содержится более 60 онкогенов, способных регулировать клеточное деление. Они находятся в неактивном состоянии в виде протоонкогенов. Под действием различных мутагенных факторов протоонкогены активируются и переходят в состояние онкогенов, которые вызывают интенсивную пролиферацию клеток и развитие опухолей. ЛЕКЦИЯ 11 Мутации числа хромосом. Гаплоидия, полиплоидия, |