МГ ответы. Медицинская генетика 104. Наследственные заболевания человека, определение, классификация, методы лабораторной диагностики. Наследственные болезни
 Скачать 1.51 Mb. Скачать 1.51 Mb.
|

|
Синдром «кошачьего крика» - частичная моносомия по короткому плечу хромосомы 5 (5р-). Синдром моносомии 5р- был первым описанным синдромом, обусловленным хромосомной мутацией (делецией). У детей с такой хромосомной аномалией отмечается необычный плач, напоминающий требовательное кошачье мяуканье или крик. По этой причине синдром сначала был назван синдромом «кошачьего крика». Частота синдрома достаточно высокая для делеционных синдромов - 1 : 45 000. Описано несколько сотен больных, поэтому цитогенетика и клиническая картина этого синдрома изучены хорошо.
Цитогенетически в большинстве случаев выявляется делеция с утратой от 1/3 до 1/2 длины короткого плеча хромосомы 5. Потеря всего короткого плеча или, наоборот, незначительного участка встречается редко. Для развития клинической картины синдрома 5р- имеет значение не величина утраченного участка, а конкретный фрагмент хромосомы. За развитие полного синдрома ответствен лишь незначительный участок в коротком плече хромосомы 5 [5р- (15,1-15,2)]. Помимо простой делеции, при этом синдроме обнаружены и другие цитогенетические варианты: кольцевая хромосома 5 (естественно, с делецией соответствующего участка короткого плеча); мозаицизм по делеции; реципрокная транслокация короткого плеча хромосомы 5 (с потерей критического участка) с другой хромосомой. Клиническая картина синдрома 5р- довольно сильно различается у отдельных больных по сочетанию врожденных пороков развития органов. Наиболее характерный признак - «кошачий крик» - обусловлен изменением гортани (сужение, мягкость хрящей, уменьшение надгортанника, необычная складчатость слизистой оболочки). Практически у всех больных имеются те или иные изменения мозговой части черепа и лица: лунообразное лицо, микроцефалия, гипертелоризм, микрогения, эпикант, антимонголоидный разрез глаз, высокое нёбо, плоская спинка носа. Ушные раковины деформированы и расположены низко. Кроме того, встречаются врожденные пороки сердца и некоторых других внутренних органов, изменения костно-мышечной системы (синдактилия стоп, клинодактилия V пальца кисти, косолапость). Выявляют мышечную гипотонию, а иногда и диастаз прямых мышц живота. Выраженность отдельных признаков и клинической картины в целом меняется с возрастом. Так, «кошачий крик», мышечная гипотония, лунообразное лицо с возрастом исчезают почти полностью, а микроцефалия выявляется более отчетливо, становятся заметнее психомоторное недоразвитие, косоглазие. Продолжительность жизни больных с синдромом 5р- зависит от тяжести врожденных пороков внутренних органов (особенно сердца), выраженности клинической картины в целом, уровня медицинской помощи и повседневной жизни. Большинство больных умирают в первые годы, около 10% больных достигают 10-летнего возраста. Имеются единичные описания больных в возрасте 50 лет и старше. 114.Синдромы анеуплоидий половых хромосом, клиническая характеристика, методы диагностики, генетический риск. Анеуплоиди́я — изменение кариотипа, при котором число хромосом в клетках не кратно гаплоидному набору (n). Отсутствие в хромосомном наборе диплоидного организма одной хромосомы называется моносомией (2n-1); отсутствие двух гомологичных хромосом — нуллисомией (2n-2); наличие дополнительной хромосомы называется трисомией (2n+1) . Анеуплоидия возникает в результате нарушения сегрегации хромосом в митозе или мейозе. Численные нарушения в системе половых хромосом (моносомия и трисомия) не вызывают таких тяжелых последствий, как аутосомные аномалии . Ярко выраженные изменения фенотипа немногочисленны или вообще отсутствуют (например, у женщин с кариотипом 47,ХХХ). В предварительной диагностике болезней, обусловленных аномалиями половых хромосом, основное значение имеет анамнез: задержка полового развития, нарушение формирования вторичных половых признаков, бесплодие, самопроизвольные аборты. Экспресс-методы цитогенетического анализа (например, определение полового хроматина в соскобе со слизистой щек) не всегда дают надежные результаты. Поэтому при подозрении на аномалию половых хромосом требуется детальное цитогенетическое исследование большого числа клеток. Поскольку серьезные анатомические дефекты отсутствуют, кариотип 47,XXY и кариотип 47,XYY y мужчин и кариотип 47,XXX у женщин часто остаются нераспознанными. Синдром Клайнфельтера (наиболее часто - 47,XXY) 1:500, 1:750 Цитогенетика: полный и мозаичный 46,XY/47,XXY варианты (мозаичность - проявления синдрома смягчены). При наличии большего числа Х-хромосом в мужском кариотипе ( кариотип 48,XXXY или кариотип 49,XXXXY ) проявления заболевания, и в том числе умственная отсталость, резко выражены . Кариотип 47,XYY вызывает менее четкую клиническую картину. Наличие Y-хромосомы определяет формирование по мужскому полу. Выявляют клинические проявления: высокий рост женский тип телосложения гинекомастия слабое оволосенение лица, подмышечных впадин, лобка отставание в физическом развитии недоразвитие половых органов и вторичных половых признаков поведенческие расстройства бесплодие (азооспермия, олигоспермия) Синдром Шерешевского - Тернера ( 45, Х) 1:2000, 1:5000 Цитогенетика многообразна: моносомия может быть в сочетании с делецией плеча Х-хромосомы, изохромосомами, кольцевыми хромасомами; частая мозаичность. Потеря Y- или второй Х-хромосомы существенно нарушают развитие организма. Если это не приводит к самопроизвольному аборту, то у новорожденных (всегда - девочек) выявляют клинические проявления: низкий рост короткая шея с избытком кожи и крыловидными , низкий рост волос на шее лимфатический отек на руках и ногах (кисти, предплечья, стопы, голени) у новорожденного гипогонадизм, недоразвитие половых органов и вторичных половых признаков (первичная аменорея, агенезия гонад, гипоплазия половых органов, слабое оволосенение лобка и подмышечных впадин, недоразвитие молочных желез, недостаточность эстрогенов) ВПР (различные почечные аномалии , сердечно-сосудистые аномалии, скелетные аномалии и эктодермальные аномалии ) нарушение скелета, черепно-лицевые дисморфии, вальгусная девиация суставов, бочкообразная грудная клетка, остеопороз. антимонголоидный разрез глазных щелей ( + птоз, эпикант) низкое расположение ушных раковин высокое небо Синдром трипло-Х (47,XXX) 1:1000 Цитогенетика: полный или мозаичный вариант Клинически проявления выражены не ярко: У большинства: нормальное физическое и психическое развитие ( 2 х-хромосомы гетерохроматизированы, функционирует лишь одна, как у нормальной женщины) нет отклонений в половом развитии, нормальная плодовитость интеллектуальное развитие на нижней границе нормы У некоторых женщин: нарушение репродуктивной функции (вторичная аменорея, дисменорея, ранняя менопауза и др.), незначительные аномалии наружных половых органов Риск: повышен риск хромосомных нарушений у потомства и возникновения спонтанных абортов Варианты Х-полисомии с числом Х-хромосом больше 3 (редко): с увеличением числа Х-хромосом нарастают отклонения от нормы – отклонения в умственном развитии, черепно-лицевые дисморфии, аномалии скелета, половых органов. Также повышен риск потомства: девочка с синдромом трипло-Х и мальчик с синдромом Кляйнфелтера Синдром дисомии по Y-хромосоме (47, ХYY) 1: 1000 У большинства: рост немного выше среднего, отклонений в физическом развитии нет поведение: дефицит внимания, гиперактивность, импульсивность нет отклонении в умственном (интеллект чуть ниже среднего), половом развитии может быть задержка речевого развития, затруднение в чтении и произношении Риск: повышенного риска иметь хромосомные аномалии у потомства НЕТ 115.Синдромы, обусловленные микрохромосомными аберрациями, клиническая характеристика, методы диагностики, генетический риск. Синдромы, обусловленные микроструктурными аберрациями хромосом ( 1 : 50 000-100 000 ) В эту группу входят синдромы, обусловленные незначительными, размером до 5 млн п.о., делециями или дупликациями строго определенных участков хромосом. Соответственно их называют микроделеционными и микродупликационными синдромами. Диагностика: современные высокоразрешающие цитогенетические методы, особенно молекулярно-цитогенетические ( метод FISH). В основе развития микроделеционных и микродупликационных синдромов лежат изменения дозы генов в участке хромосомы, затронутом перестройкой. Для формирования клинической картины данной группы заболеваний принципиально важно отсутствие продукта нескольких генов, затрагиваемых микроделецией. По своей природе смежные генные синдромы находятся на границе между менделирующими моногенными заболеваниями и хромосомными болезнями. Патологический процесс при некоторых из них развертывается через инактивацию опухолесупрессоров (ретинобластомы, опухоли Вильмса), клиника других синдромов обусловлена не только делециями как таковыми, но и явлениями хромосомного импринтинга и одно-родительских дисомий (синдромы Прадера-Вилли, Ангельмана, Беквита-Видемана).  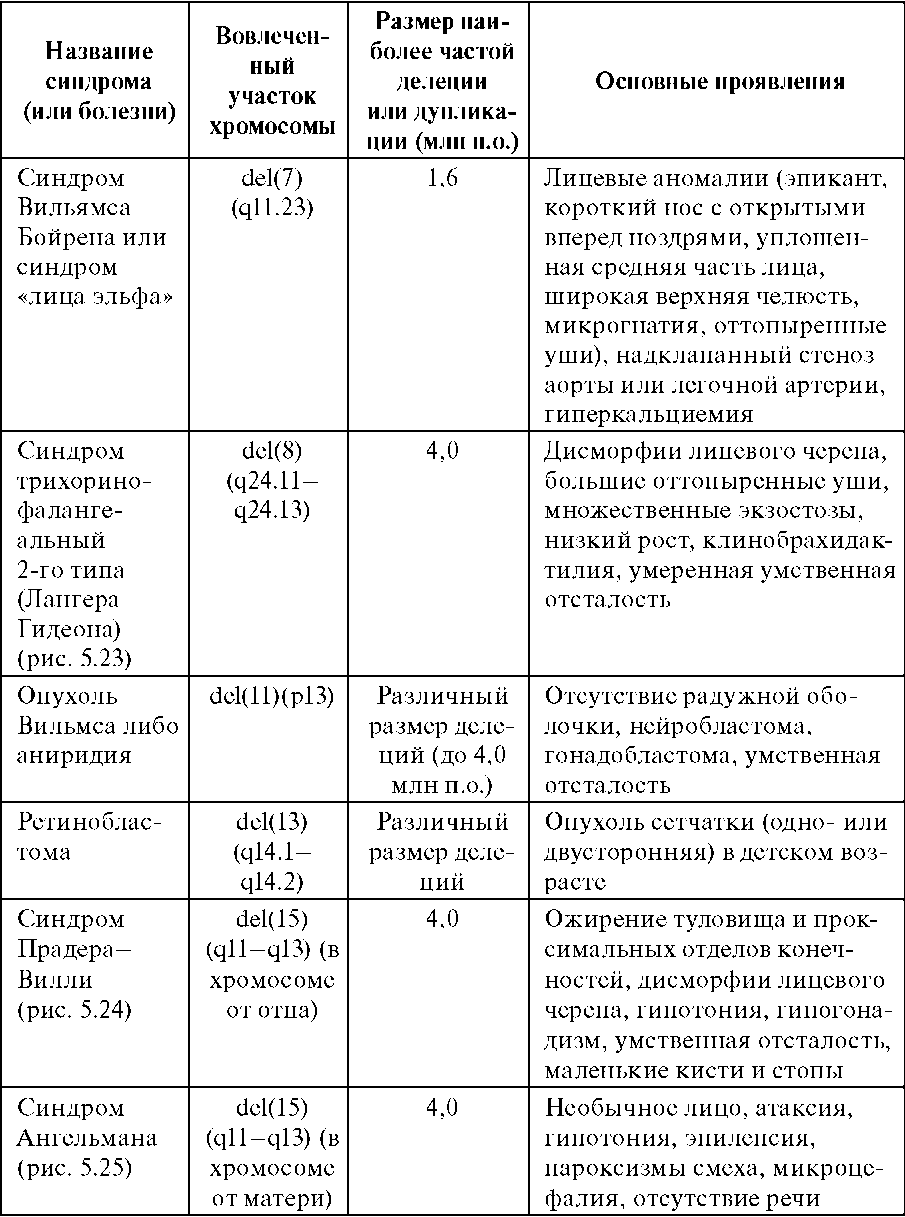 Клинические проявления синдромов сильно варьируют в связи с разной протяженностью делеции или дупликации, а также в связи с родительской принадлежностью микроперестройки - унаследована ли она от отца или от матери. В последнем случае речь идет об импринтинге на хромосомном уровне. Это явление было открыто при цитогенетическом изучении двух клинически различающихся синдромов (Прадера-Вилли и Ангельмана). В обоих случаях микроделеция наблюдается в хромосоме 15 (участок q11-q13). 116.Генные болезни человека, классификации, современные методы лабораторной диагностики. Генные болезни - разнородная по клиническим проявлениям группа заболеваний, обусловленных генными мутациями. Основа для их объединения - этиологическая генетическая характеристика и закономерности наследования в семьях и популяциях. У человека описаны все типы генных мутаций,обусловливающие наследственные болезни: миссенс, нонсенс, сдвиг рамки считывания, делеции, вставки (инсерции), нарушения сплайсинга, увеличение числа (экспансия) тринуклеотидных повторов. Любой из этих видов мутаций может вести к наследственным болезням. Даже одна и та же генная болезнь может быть обусловлена разными мутациями одного и того же гена. Классификация: Генетическая классификация: согласно типам наследования: аутосомно-доминантные (нейрофиброматоз I типа, синдром Марфана) аутосомно-рецессивные ( фенилкетонурия, галактоземия, болезнь Вильсона - Коновалова) Х-сцепленные доминантные (витамин Д резистентный рахит) Х-сцепленные рецессивные (миопатия Дюшенна-Беккера, гемофилия, синдром Леша-Хантера) Y-сцепленные (голандрические) митохондриальные (атрофия зрительного нерва Лебера, митохондриальные миопатии) – передаются только от матери Отнесение болезни к той или иной группе помогает врачу сориентироваться относительно ситуации в семье и определить вид медико-генетической помощи. Клиническая классификация: систему или орган, наиболее вовлеченный в патологический процесс: нервные (нейрофиброматоз) нервно-мышечные (миотоническая дистрофия, миодистрофия Дюшенна-Беккера) эндокринные (адреногенитальный синдром) кожные, глазные, опорно-двигательного аппарата, крови, сердечно-сосудистой системы, психические, мочеполовой системы, желудочно-кишечного тракта (ЖКТ), легких. Для некоторых групп болезней установились даже специальные термины: нейрогенетика, дерматогенетика, офтальмогенетика. Условность клинического принципа классификации очевидна. Некоторые болезни у одних больных больше проявляются в одной системе, у других - в другой. Например, муковисцидоз может преимущественно поражать или ЖКТ, или легкие. Нейрофиброматоз I типа может выражаться либо кожными изменениями (пигментные пятна, нейрофибромы),либо опухолями нервных стволов и мозга. Патогенетическая классификация: основное патогенетическое звено. В соответствии с этим различают Наследственные болезни обмена веществ углеводного обмена(галактоземия, гликогенозы) аминокислотного обмена (фенилкетонурия) обмен витаминов (витамин-резистентный рахит) обмена липидов (семейная гиперхолестеринемия) обмена металлов и др Врожденные пороки развития (моногенной природы) Комбинированные состояния Лабораторная диагностика: Молекулярно – генетические методы (методы ДНК-диагностики – прямые и косвенные) Биохимические методы – выявление биохимического фенотипа организма хроматографические методы анализа масс-спектрометрия тандемная масс-спектрометрия и др. 117.Синдром Марфана и Элерса-Данло, клиническая характеристика, методы диагностики, генетический риск. Синдром Марфана (1 : 10 000-15 000) Синдром Марфана - наследственная аутосомно-доминантная болезнь соединительной ткани. Причина: мутации в гене фибриллина (локализация в хромосоме 15q21) - в основном миссенс. Симптоматика синдрома Марфана многосистемная и разнообразная: от легких форм, трудноотличимых от нормы, до инвалидизирующего течения. Диагностика: молекулярно – генетические методы (методы ДНК-диагностики – прямые и косвенные) + клинический анализ по критериям Наиболее специфичны для синдрома Марфана нарушения скелета, вывих хрусталика, сердечно-сосудистые изменения, эктазия твердой мозговой оболочки. -Мышечно-скелетная система: арахнодактилия, долихостеномелия, высокий рост, длинные конечности, деформация позвоночника (сколиоз, грудной лордоз, гиперкифоз), деформация передней стенки грудной клетки (вдавленная грудь, «куриная» грудь или оба варианта), ненормальная подвижность суставов (гиперподвижность, врожденные контрактуры или оба варианта), плоская стопа, высокое арковидное нёбо, недоразвитие вертлужной впадины, мышечная гипотония 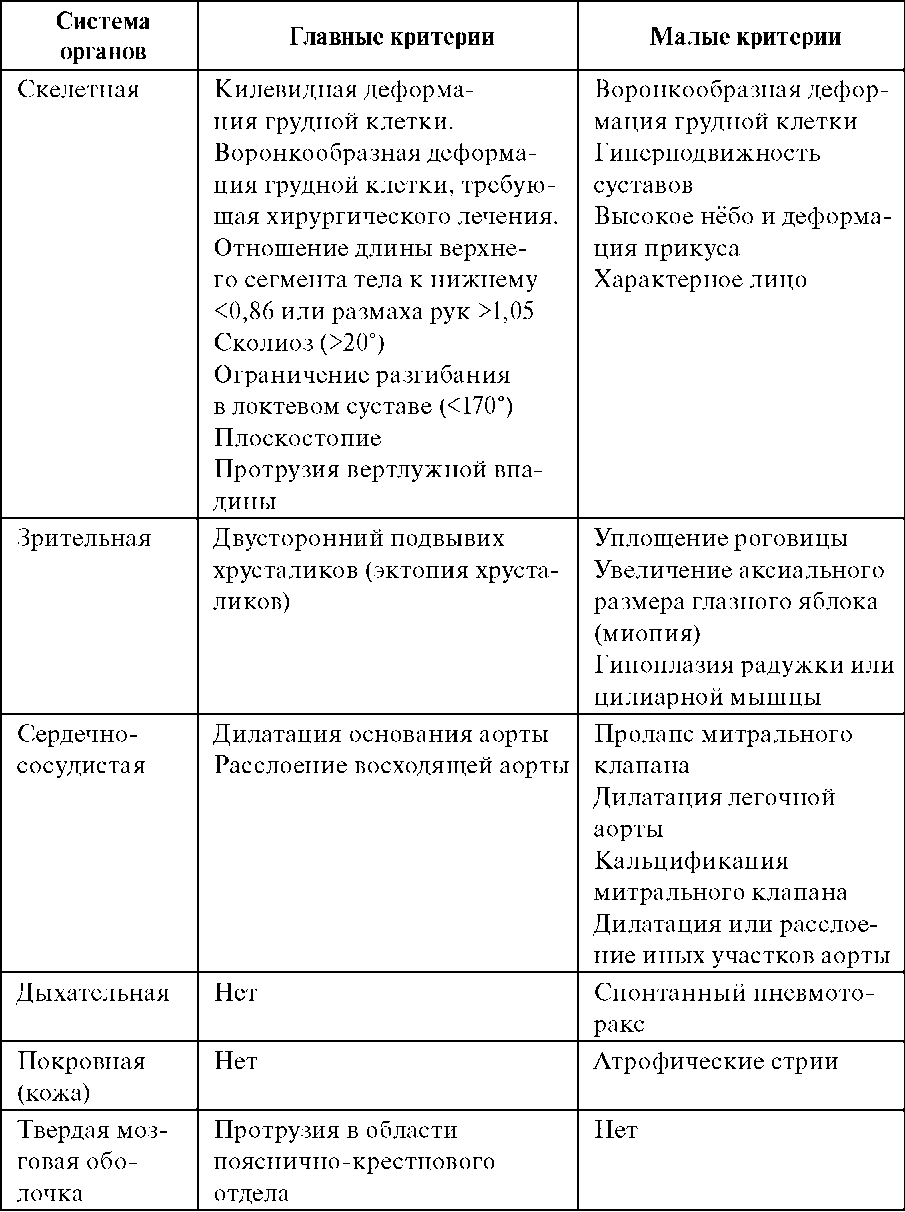  Диагноз: по 1 главному критерию в двух системах и одного малого - в третьей системе органов. При синдроме Марфана ростовые скачки и закрытие зон роста скелета наблюдаются на 2,4 года раньше у лиц мужского пола и на 2,2 года раньше у лиц женского пола. Рост взрослых мужчин равен в среднем 191 см, женщин - 175 см. С увеличением возраста отца (особенно после 35 лет) повышается вероятность рождения ребенка с синдромом Марфана. Синдром Элерса-Данло Синдром Элерса-Данло - гетерогенная группа наследственных болезней соединительной ткани с разными типами наследования (аутосомно-доминантный, аутосомной – рецессивный, рецессивный Х-сцепленный) Диагностика: молекулярно – генетические методы (методы ДНК-диагностики – прямые и косвенные) + клиническая картина Синдром Элерса-Данло проявляется врожденной гиперрастяжимостью соединительной ткани в связи с нарушениями синтеза коллагена, обусловленными мутациями в разных генах коллагена и других белков экстраклеточного матрикса. Клинически, биохимически, молекулярно-генетически идентифицировано 6 типов синдрома Элерса-Данлоетные изменения, повышенная ранимость кожи, проявления со стороны внутренних органов. - Кожа: Сверхрастяжимость (щеки, под наружными концами ключиц, локти, колени), бархатистость, хрупкость, кровоточивость, темно-коричневые веснушки (более 20), рубцы (множественные, типа папиросной бумаги, келоидные), стрии в области поясницы, просвечивающие вены, расхождение послеоперационных швов - Суставы: пассивное разгибание мизинца на 90° и более, приведение большого пальца кисти к предплечью, переразгибание локтевого сустава на 10° и более, переразгибание коленного сустава на 10° и более, свободное касание ладонями пола при несогнутых коленях, переразгибание межфаланговых, запястных, голеностопных и других суставов, привычный вывих суставов, плоскостопие - Глаза: птоз, избыточное развитие периорбитальной клетчатки, отслойка сетчатки, остатки эпиканта, разрыв глазного яблока. - Уши: сверхрастяжимость. - Зубы: частичная адентия, сверхкомплектные зубы, опалесцирующая эмаль, пародонтоз, множественный кариес. - Грудная клетка: сколиоз, кифоз, лордоз, плоская спина, вдавление грудины. - Живот: грыжи (пупочная, белой линии, паховая, диафрагмальная), спонтанная перфорация кишечника. - Конечности: варикозные вены, подкожные подвижные узелки на голенях, плоскостопие. - Сердце: пролапс митрального клапана, аритмии, вегетососудистая дисто-ния. - Внутренние органы: птоз желудка, почек и матки. - Мозг: аневризмы сосудов мозга, субарахноидальное кровоизлияние. - Стремительные роды. Наличие синдрома Элерса- Данло мало отражается на репродуктивной функции, хотя у больных снижено количество потомков. При синдроме Элерса- Данло имеются нарушения (первичные или вторичные) во всех системах организма. Наиболее важные диагностические признаки: гиперэластичность кожи, подкожные узелки (сферулы), легче прощупываемые на передней поверхности голени; переразгибание суставов; повышенная ранимость тканей; симптомы геморрагического диатеза; пролапс митрального клапана. Классификация (6 типов): |