МГ ответы. Медицинская генетика 104. Наследственные заболевания человека, определение, классификация, методы лабораторной диагностики. Наследственные болезни
 Скачать 1.51 Mb. Скачать 1.51 Mb.
|

|
110.Методы ДНК-диагностики наследственной патологии. С помощью прямых методов выявляются нарушения в первичной нуклеотидной последовательности ДНК (мутации и их типы). Прямые методы отличаются точностью, достигающей почти 100 %. Однако на практике указанные методы могут применяться при определенных условиях: 1) известной цитогенетической локализации гена, ответственного за развитие наследственного заболевания, 2) должен быть клонированным ген заболевания и известна его нуклеотидная последовательность. Целью прямой диагностики является идентификация мутантных аллелей (нарушения в первичной нуклеотидной последовательности ДНК, мутации и их типы). Высокая точность метода прямой ДНК-диагностики в большинстве случаев не требует ДНК-анализа всех членов семьи, так как выявление мутации в соответствующем гене позволяет почти со 100-процентной точностью подтвердить диагноз и определить генотип всех членов семьи больного ребенка, включая гетерозиготных носителей. Недостатком метода прямой ДНК-диагностики является необходимость знания точной локализации гена и спектра его мутаций. Методы прямой ДНК-диагностики показаны для таких заболеваний, как фенилкетонурия (мутация R408W), муковисцидоз - (наиболее частая мутация delF508), хорея Гентингтона (экспансия тринуклеотидных повторов-CTG-повторы) и др. Однако к настоящему времени гены многих заболеваний не картированы, неизвестна их экзонно-интронная организация, и многие наследственные болезни отличаются выраженной генетической гетерогенностью, что не позволяет в полной мере использовать прямые методы ДНК-диагностики. Поэтому информативность метода прямой ДНК-диагностики широко варьирует. В связи с этим используются косвенные методы молекулярно-генетической диагностики наследственных болезней. Косвенные методы ДНК-диагностики основаны на анализе сцепления с исследуемым геном определенного полиморфного локуса (маркера), с помощью которого можно производить маркировку как мутантных, так и нормальных аллелей и проанализировать их передачу в поколениях, т.е. среди родственников обследуемого лица. Это особенно важно при решении вопроса о пренатальной (дородовой) диагностике наследственного заболевания. При использовании косвенных методов ДНК-диагностики следует помнить — чем теснее сцепление между маркерным локусом и мутантным геном, тем точнее диагноз. Чтобы свести до минимума ошибку диагностики, необходимо по возможности использовать внутригенные маркеры или использовать два маркерных локуса, фланкирующих мутантный аллель. Таким образом, косвенная ДНК-диагностика проводится в следующих случаях: 1) когда ген не идентифицирован, а лишь картирован на определенной хромосоме, 2) когда методы прямой ДНК-диагностики не дают результата (например, в силу большой протяженности гена или широком спектре мутационных изменений, 3) при сложной экзонно-интронной организации гена. При использовании косвенных методов ДНК-диагностики требуется семейный анализ аллелей полиморфных маркеров. Для косвенной диагностики могут использоваться так называемые гипервариабельные сателлитные повторы. Косвенные методы ДНК-диагностики могут использоваться в пренатальной диагностике практически для всех моногенных заболеваний. Однако для этого необходимо иметь знания о том, что локус является высокополиморфным и находится вблизи от мутантного гена или внутри него. Поэтому для диагностики требуется обследование как можно большего числа родственников (в первую очередь родители—дети), чтобы проследить путь передачи маркеров потомству. Это повышает информативность выбранного маркера. 111.Хромосомные болезни человека, характерные клинические признаки, классификация, методы диагностики. Хромосомные болезни — наследственные заболевания, обусловленные изменением числа или структуры хромосом. К хромосомным относятся болезни, обусловленные геномными мутациями или структурными изменениями отдельных хромосом. Хромосомные болезни возникают в результате мутаций в половых клетках одного из родителей. Из поколения в поколение передаются не более 3—5 % из них. Хромосомными нарушениями обусловлены примерно 50 % спонтанных абортов и 7 % всех мёртворождений. Все хромосомные болезни принято делить на две группы: аномалии числа хромосом и нарушения структуры хромосом. Аномалии числа хромосом Болезни, обусловленные нарушением числа хромосом синдром Дауна — трисомия по 21 хромосоме, к признакам относятся: слабоумие, задержка роста, характерная внешность, изменения дерматоглифики; синдром Патау — трисомия по 13 хромосоме, характеризуется множественными пороками развития, идиотией, часто — полидактилия, нарушения строения половых органов, глухота; практически все больные не доживают до одного года; синдром Эдвардса — трисомия по 18 хромосоме, нижняя челюсть и ротовое отверстие маленькие, глазные щели узкие и короткие, ушные раковины деформированы; 60% детей умирают в возрасте до 3-х месяцев, до года доживают лишь 10%, основной причиной служит остановка дыхания и нарушение работы сердца. Болезни, связанные с нарушением числа половых хромосом Синдром Шерешевского — Тёрнера — отсутствие одной Х-хромосомы у женщин (45 ХО) вследствие нарушения расхождения половых хромосом; к признакам относится низкорослость, половой инфантилизм и бесплодие, различные соматические нарушения (микрогнатия, короткая шея и др.); полисомия по Х-хромосоме — включает трисомию (кариотии 47, XXX), тетрасомию (48, ХХХХ), пентасомию (49, ХХХХХ), отмечается незначительное снижениеинтеллекта, повышенная вероятность развития психозов и шизофрении с неблагоприятным типом течения; полисомия по Y-хромосоме — как и полисомия по X-хромосоме, включает трисомию (кариотии 47, XYY), тетрасомию (48, ХYYY), пентасомию (49, ХYYYY), клинические проявления также схожи с полисомией X-хромосомы; Синдром Клайнфельтера — полисомия по X- и Y-хромосомам у мальчиков (47, XXY; 48, XXYY и др.), признаки: евнухоидный тип сложения, гинекомастия, слабый рост волос на лице, в подмышечных впадинах и на лобке, половой инфантилизм, бесплодие; умственное развитие отстает, однако иногда интеллект нормальный. Болезни, причиной которых является полиплоидия триплоидии, тетраплоидии и т. д.; причина — нарушение процесса мейоза вследствие мутации, в результате чего дочерняя половая клетка получает вместо гаплоидного (23) диплоидный (46) набор хромосом, то есть 69 хромосом (у мужчин кариотип 69, XYY, у женщин — 69, XXX); почти всегда летальны до рождения. Нарушения структуры хромосом Транслокации — обменные перестройки между негомологичными хромосомами. Делеции — потери участка хромосомы. Например, синдром кошачьего крика связан с делецией короткого плеча 5-й хромосомы. Признаком его служит необычный плач детей, напоминающий мяуканье или крик кошки. Это связано с патологией гортани или голосовых связок. Наиболее типичным, помимо «кошачьего крика», является умственное и физическое недоразвитие, микроцефалия (аномально уменьшенная голова). Инверсии — повороты участка хромосомы на 180 градусов. Дупликации — удвоения участка хромосомы. Изохромосомия — хромосомы с повторяющимся генетическим материалом в обоих плечах. Возникновение кольцевых хромосом — соединение двух концевых делеций в обоих плечах хромосомы. Какова же общая клиническая характеристика хромосомных болезней? Почти все они сопровождаются множественными нарушениями скелета, психики. Отмечаются врожденные пороки наружных и внутренних половых органов, их замедленный рост. Нарушается деятельность нервной, эндокринной и других систем, снижена генеративная функция, наблюдается четкое повышение смертности среди лиц с хромосомными аномалиями. Диагностические признаки разделяются на 3 группы. А) комплекс признаков, позволяющих лишь заподозрить хромосомную аномалию. Это общие признаки: физическое недоразвитие, ряд дизморфий мозгового и лицевого черепа (деформация ушных раковин и их низкое расположение, микроцефалия, эпикант, высокое небо), косолапость, клинодактилия мизинцев, некоторые пороки развития внутренних органов (сердца, почек, легких). В) признаки встречаются в основном при определенных хромосомных болезнях. Их сочетание позволяет в большинстве случаев диагностировать хромосомную аномалию. Среди характерных, наиболее часто встречающихся признаков этой группы при трисомии хромосомы 18 следует назвать долихоцефалию (89,6% случаев), флексорное положение кистей (96,1 %), «стопу-качалку» (76,2%), короткий и широкий I палец стопы (70,6% случаев); при трисомии по хромосоме 13--расщелину верхней губы и неба (68,7 % случаев), флексорное положение кистей (44,4%), косоглазие (31,4%), дефект скальпа (30,5 % случаев) и др. С) признаки характерны только для одной хромосомной аномалии, например, «кошачий крик» -- при синдроме 5р--, алопеция при синдроме 18р. Хромосомным болезням свойственна чрезмерная фенотипическая (клиническая) вариабельность. Часто при одних и тех же хромосомных аномалиях клинические признаки выражены по-разному. В качестве примера можно привести болезнь Дауна, при которой поражение психики проявляется слабоумием от легких до тяжелых степеней (дебильность -- имбецильность -- идиотия). Выраженность клинических проявлений хромосомных болезней зависит от многих причин, среди которых следует отметить генотипические и паратипические факторы, состав поражаемых генов, размер аберрации и индивидуальность хромосомы, процент мозаичных клеток в организме и т.д. Для диагностики хромосомных болезней в настоящее время применяют ряд методов медицинской генетики, чаще клинико-генеалогический, цитогенетический (определение полового хроматина и кариотипирование), патологоанатомический и дерматоглифический. Как правило, современная диагностика любого заболевания является комплексной. Кроме традиционных клинических данных, лабораторных исследований, сбора анемнестических данных, при диагностике наследственных болезней, в частности хромосомных, особое внимание уделяется изучению генеалогии больного. Только около 3-5% их четко наследуется. 112.Особенности клинических проявлений хромосомных синдромов, обусловленных трисомиями аутосом. Методы диагностики, генетический риск. Синдром Дауна – трисомия 21 Врожденные пороки развития, нарушения постнатального развития нервной системы, и вторичный иммунодефицит и т.п. Дети с синдромом Дауна рождаются в срок, но с умеренно выраженной пренатальной гипоплазией (на 8-10% ниже средних величин). монголоидный разрез глаз (по этой причине синдром Дауна долго называли монголоидизмом), брахицефалия, круглое уплощенное лицо, плоская спинка носа, эпикант, крупный (обычно высунутый) язык, деформированные ушные раковины. мышечная гипотония сочетается с разболтанностью суставов. ВПР клинодактилия, типичные изменения дерматоглифики [четырехпальцевая, или «обезьянья», складка на ладони, две кожные складки вместо трех на мизинце, высокое положение трирадиуса и др.]. Врожденные пороки внутренних органов, сниженная приспособляемость детей с синдромом Дауна часто приводят к смерти в первые 5 лет. Следствием измененного иммунитета и недостаточности репарационных систем (для поврежденной ДНК) являются лейкозы, часто возникающие у больных с синдромом Дауна. Синдром Эдвардса - трисомия 18 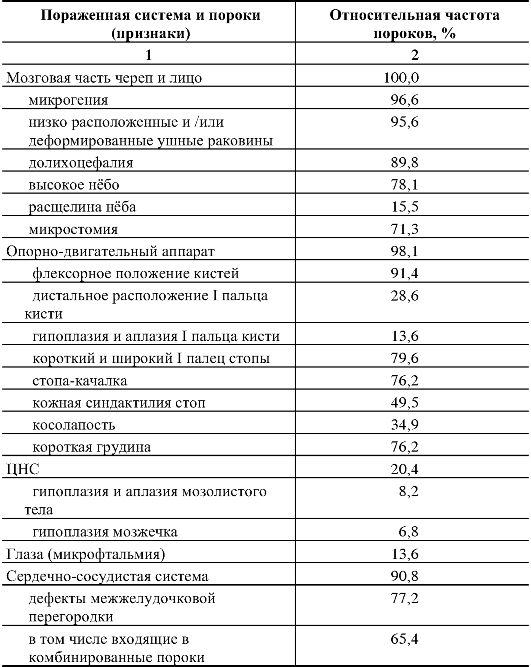 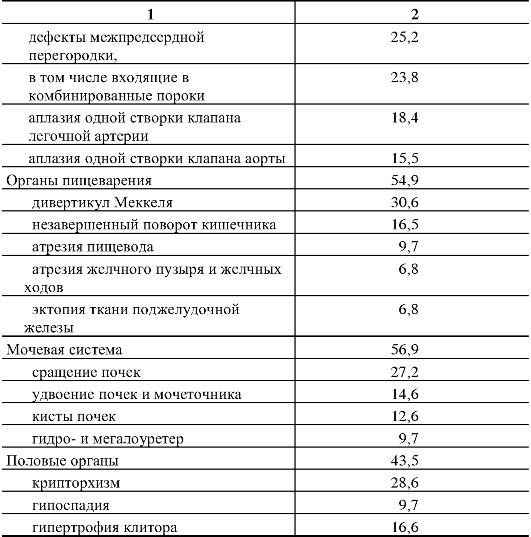 Наиболее значимы в диагностике синдрома Эдвардса изменения мозгового черепа и лица, опорно-двигательного аппарата, пороки развития сердечно-сосудистой системы. Дети с синдромом Эдвардса умирают в раннем возрасте (90% до 1 года) от осложнений, обусловленных врожденными пороками развития (асфиксия, пневмония, кишечная непроходимость, сердечнососудистая недостаточность). Клиническая и даже патологоанатомическая дифференциальная диагностика синдрома Эдвардса сложна, поэтому во всех случаях показано цитогенетическое исследование. Показания для него те же, что и при трисомии 13. Синдром Патау - трисомия 13 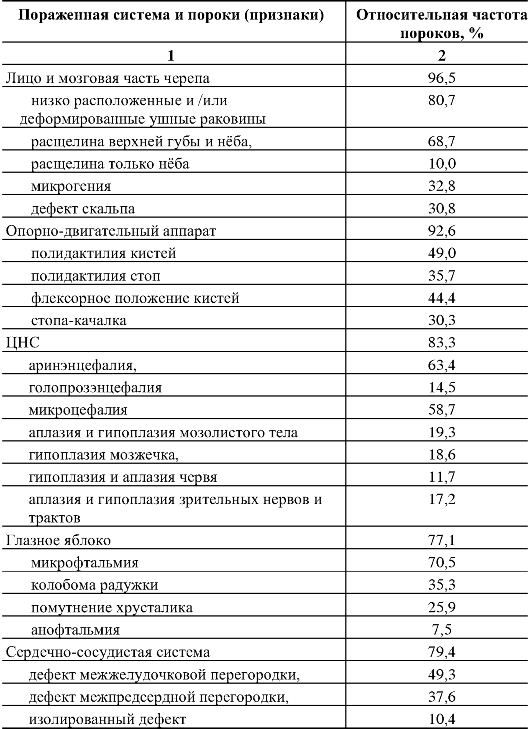 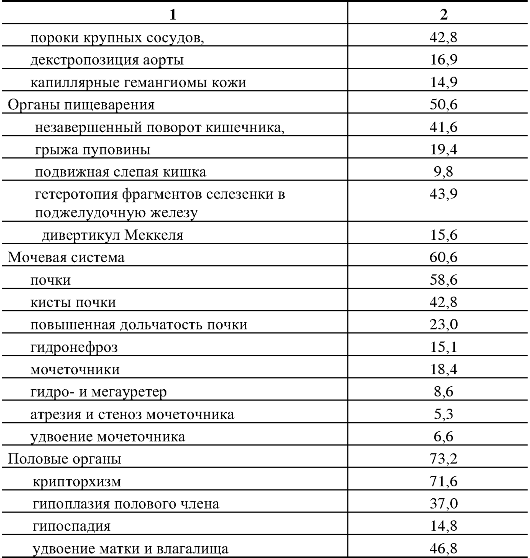 Клиническая диагностика синдрома Патау основывается на сочетании характерных пороков развития. При подозрении на синдром Патау показано УЗИ всех внутренних органов. В связи с тяжелыми врожденными пороками развития большинство детей с синдромом Патау умирают в первые недели или месяцы жизни (95% умирают до 1 года). Однако некоторые больные живут несколько лет. Более того, в развитых странах отмечается тенденция увеличения продолжительности жизни больных с синдромом Патау до 5 лет (около 15% больных ) и даже до 10 лет (2-3% больных). Трисомия 8 Для болезни наиболее характерны отклонения в строении лица, пороки опорно-двигательного аппарата и мочевой системы. Это выступающий лоб, косоглазие, эпикант, глубоко посаженные глаза, гипертелоризм глаз и сосков, высокое нёбо (иногда расщелина), толстые губы, вывернутая нижняя губа, большие ушные раковины с толстой мочкой, контрактуры суставов, камптодактилия, аплазия надколенника, глубокие борозды между межпальцевыми подушечками, четырехпальцевая складка, аномалии ануса. При УЗИ выявляются аномалии позвоночника (добавочные позвонки, неполное закрытие позвоночного канала), аномалии формы и положения ребер или добавочные ребра. При трисомии 8 прогноз физического, психического развития и жизни неблагоприятный, хотя описаны пациенты в возрасте 17 лет. Со временем у больных проявляются умственная отсталость, гидроцефалия, паховая грыжа, новые контрактуры, аплазия мозолистого тела, кифоз, сколиоз, аномалии тазобедренного сустава, узкий таз, узкие плечи. 113.Особенности клинических проявлений хромосомных синдромов, обусловленных хромосомными аберрациями. Методы диагностики, генетический риск. Различные хромосомные аберрации встречаются с разной частотой. По сводным данным многих исследований, распространенность наиболее частых хромосомных аберраций среди новорожденных следующая: 21-трисомия (синдром Дауна) - 1:700; XXX (трисомия Х) - 1:1000 (девочки); XYY (синдром дубль-Y) - 1:1000 (мальчики); XXY (синдром Клайнфельтера) - 1:1400 (мальчики); ХО (синдром Шерешевского - Тернера) - 1:3300 (девочки); 5р (синдром «кошачьего крика») - 1:4000; 18-трисомия (синдром Эдвардса) - 1:6800; 13-трисомия (синдром Патау) - 1:7600. Наиболее часто встречаются изменения модального числа хромосом. Это отсутствие в хромосомном наборе какой-либо хромосомы (моносомия) или появление добавочной хромосомы (трисомия, тетрасомия и т. д.). Примером таких аномалий являются хорошо известные клиницистам четко очерченные клинические синдромы - синдром Дауна (21-трисомия), синдром Эдвардса (18-трисомия), синдром Патау (13-трисомия), синдром Клайнфелтера (XXY), синдром Шерешевского - Тернера (ХО). К другим хромосомным аберрациям относятся такие нарушения, при которых общее число хромосом может оставаться нормальным, а изменяется структура самой хромосомы: транслокации (обмен сегментами между хромосомами), делеции (отсутствие части хромосомы), кольцевые хромосомы и т. д. Перспективным методом профилактики хромосомной патологии является антенатальная диагностика, т. е. исследование клеток амниотической жидкости на 16-18-й неделе беременности или клеток хориона в более ранние сроки. Внедрение антенатальной диагностики в широкую практику с охватом контингентов повышенного риска позволит значительно сократить частоту рождения детей с хромосомными заболеваниями, в первую очередь с болезнью Дауна. Синдром Шерешевского - Тернера ( 45, Х) 1:2000, 1:5000 Цитогенетика многообразна: моносомия может быть в сочетании с делецией плеча Х-хромосомы, изохромосомами, кольцевыми хромасомами; частая мозаичность. Потеря Y- или второй Х-хромосомы существенно нарушают развитие организма. Если это не приводит к самопроизвольному аборту, то у новорожденных (всегда - девочек) выявляют клинические проявления: низкий рост короткая шея с избытком кожи и крыловидными , низкий рост волос на шее лимфатический отек на руках и ногах (кисти, предплечья, стопы, голени) у новорожденного гипогонадизм, недоразвитие половых органов и вторичных половых признаков (первичная аменорея, агенезия гонад, гипоплазия половых органов, слабое оволосенение лобка и подмышечных впадин, недоразвитие молочных желез, недостаточность эстрогенов) ВПР (различные почечные аномалии , сердечно-сосудистые аномалии, скелетные аномалии и эктодермальные аномалии ) нарушение скелета, черепно-лицевые дисморфии, вальгусная девиация суставов, бочкообразная грудная клетка, остеопороз. антимонголоидный разрез глазных щелей ( + птоз, эпикант) низкое расположение ушных раковин высокое небо Рентгенологическом исследовании отмечаются задержка окостенения, нарушение слияния эпифизов с метафазами, остеопороз трубчатых костей. На электроэнцефалограмме нередко наблюдаются признаки задержки коркового электрогенеза, дизритмия. Умственное недоразвитие выявляется у незначительной части больных: чаще оно выражено нерезко, но изредка достигает степени имбецильности. Обычно больные трудолюбивы и благодушны. Описан своеобразный инфантилизм со склонностью к домовитости, стремлением опекать и поучать младших. У многих больных с возрастом появляются критика к своему состоянию и переживание дефекта: они становятся более замкнутыми, раздражительными, склонны к невротическим реакциям. Диагноз может быть заподозрен клинически и окончательно ставится при цитогенетическом исследовании. В типичных случаях в хромосомном наборе больных выявляется 45 хромосом (45/ХО) - 22 пары аутосом и только одна Х-хромосома. Синдром Клайнфельтера (наиболее часто - 47,XXY) 1:500, 1:750 Цитогенетика: полный и мозаичный 46,XY/47,XXY варианты (мозаичность - проявления синдрома смягчены). При наличии большего числа Х-хромосом в мужском кариотипе ( кариотип 48,XXXY или кариотип 49,XXXXY ) проявления заболевания, и в том числе умственная отсталость, резко выражены . Кариотип 47,XYY вызывает менее четкую клиническую картину. Наличие Y-хромосомы определяет формирование по мужскому полу. Выявляют клинические проявления: высокий рост женский тип телосложения гинекомастия слабое оволосенение лица, подмышечных впадин, лобка отставание в физическом развитии недоразвитие половых органов и вторичных половых признаков поведенческие расстройства бесплодие (азооспермия, олигоспермия) У этих больных наряду с недостаточностью внимания, восприятия, памяти и абстрактного мышления более резко и рельефно обнаруживаются чрезмерная внушаемость, подражательность, подчиняемость, несамостоятельность, чрезмерная привязанность к близким, нередко с элементом назойливости. Настроение обычно повышенное, с эйфорическим оттенком, имеет тенденцию к беспричинным колебаниям, иногда отмечается склонность к эксплозивным аффективным вспышкам. У этих больных выявляются недостаточное чувство долга, ответственности, активности, а также неспособность к длительному волевому усилию и напряженной деятельности. Эти особенности эмоционально-волевой сферы как бы выступают на первый план в структуре дефекта и утяжеляют общую клиническую картину психического недоразвития. При легкой форме психического недоразвития с началом обучения в школе и особенно в пубертатном и постпубертатном возрасте у больных часто появляется сознание своей неполноценности, которое становится источником внутреннего конфликта. Начинает преобладать гипотимный фон настроения, нередко с раздражительностью, легко возникают невротические и патохарактерологические реакции. В литературе также описываются случаи синдрома Клайнфелтера с депрессивными, ипохондрическими, навязчивыми, нарколептическими и шизофреноподобными расстройствами. Окончательный диагноз основывается на цитогенетическом исследовании: обнаруживают в ядрах клеток высокое содержание полового хроматина, соответствующее женскому типу. Кариологическое исследование выявляет 47 хромосом (47, XXY). |