|
|
шпоры по бх-последняя итоговая. Основные системы межклеточной коммуникации эндокринная, паракринная, аутокринная регуляция
Хроническое воспаление – увеличение содержания γ-глобулинов(ревматоидный артрит, хронический гепатит).Нефротический синдром – повышение концентрации в крови α-2-глобулинов(происходит за счет накопленияальфа-2-макроглобулинана фоне потери альбумина и других белков при фильтрации в почечных клубочках).Цирроз печени – значительное увеличение белков гамма-фракции.Диспротеинемия — нарушение нормального соотношения между фракциями белков; наблюдается при болезнях печени (острый и хронический гепатит, цирроз), почек (острый и хронический гломерулонефрит, амилоидоз, нефротический синдром), крови (лейкоз), воспалительных и аллергических заболеваниях, ревматизме, инфаркте миокарда.
Энзимодиагностика: механизмы изменения уровня активности ферментов в крови
Энзимодиагностика заключается в постановке диагноза заболевания (или синдрома) на основе определения активности ферментов в биологических жидкостях человека. Принципы энзимодиагностики основаны на следующих позициях:
при повреждении клеток в крови или других биологических жидкостях (например, в моче) увеличивается концентрация внутриклеточных ферментов повреждённых клеток; количество высвобождаемого фермента достаточно для его обнаружения;
активность ферментов в биологических жидкостях, обнаруживаемых при повреждении клеток, стабильна в течение достаточно длительного времени И отличается от нормальных значений; ряд ферментов имеет преимущественную или абсолютную локализацию в определённых органах (органоспецифичность); существуют различия во внутриклеточной локализации ряда ферментов.
При многих заболеваниях происходит повреждение клеток, и их содержимое, в том числе и ферменты, высвобождаются в кровь. К причинам, вызывающим высвобождение внутриклеточного содержимого в кровь, относят нарушение проницаемости мембраны клеток (при воспалительных процессах) или нарушение целостности клеток (при некрозе). Определение в крови активности ряда ферментов хорошо налажено в биохимических лабораториях, что используют для диагностики заболеваний сердца, печени, скелетной
мускулатуры и других тканей. Уровень активности ферментов в плазме коррелирует со степенью повреждения клеток.
Для энзимодиагностики имеют большое значение знания о субклеточной локализации ферментов. Так, появление в плазме крови ферментов, имеющих только цитозольную локализацию, свидетельствует о воспалительном процессе; при обнаружении митохондриальных или ядерных ферментов можно говорить о более глубоких повреждениях клетки, например о некрозе.
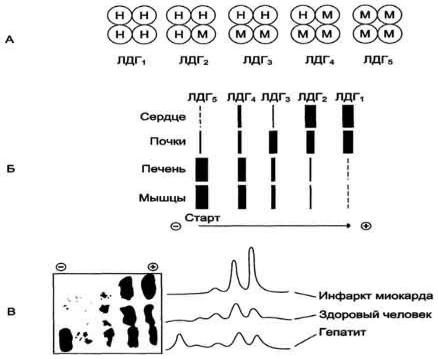 . .
2. Изоферменты
Ферменты, катализирующие одну и ту же химическую реакцию, но отличающиеся по первичной структуре белка, называют изофермен-тами,или изоэнзимами. Они катализируют один и тот же тип реакции с принципиально одинаковым механизмом, но отличаются друг от друга кинетическими параметрами, условиями активации, особенностями связи апофермента и кофермента. Появление в эволюции различных изоформ ЛДГ обусловлено особенностями окислительного метаболизма тканей. Изоферменты ЛДГ4 и ЛДГ5 (М-типыЛДГ) работают эффективно в анаэробных условиях, ЛДГ, и ЛДГ2(Н-типы)- в аэробных, когда пируват быстро окисляется до СО2 и Н2О, а не восстанавливается до молочной кислоты.
При ряде заболеваний исследуют активность ЛДГ в плазме крови. В норме активность ЛДГ составляет 170520 ЕД/л. Повышение активности наблюдают при острых поражениях сердца, печени, почек, а также при мегалобластных и гемолитических анемиях. Однако это указывает на повреждение лишь одной из перечисленных тканей.
Для постановки диагноза необходимо исследование изоформ ЛДГ в плазме крови методом электрофореза.
Изоформы креатинкиназы. Креатинкиназа (КК) катализирует реакцию образования креатинфосфата: Молекула КК - димер, состоящий из субъединиц двух типов: М и В . Из этих субъединиц образуются 3 изофермента - ВВ, MB, MM. Изофермент ВВ находится преимущественно в головном мозге, ММ - в скелетных мышцах и MB - в сердечной мышце. Изоформы КК имеют разную электрофоретическую подвижность .
Активность КК в норме не должна превышать 90 МЕ/л. Определение активности КК в плазме крови имеет диагностическое значение при инфаркте миокарда (происходит повышение уровня МВ-изоформы).Количество изоформы ММ может повышаться при травмах и повреждениях скелетных мышц. Изоформа ВВ не может проникнуть через гематоэнцефалический барьер, поэтому в крови практически не определяется даже при инсультах и диагностического значения не имеет.
|
Энзимодиагностика при инфаркте миокарда и заболеваниях печени.
При инфаркте миокарда наблюдают достоверные изменения в крови активности ферментов КК, ЛДГ и аспартатаминотрансферазы - ACT, которые зависят от времени, прошедшего от начала развития инфаркта и от зоны тканевого повреждения
ККизоформы MB, однако фермент быстро удаляется из кровотока. Обнаружение повышенной активности
ККв плазме крови - основной энзимодиагностический критерий инфаркта миокарда. Если у пациента с загрудинными болями не обнаружено изменения в активности КК, диагноз инфаркта миокарда маловероятен. Дополнительным подтверждением диагноза инфаркта миокарда служит обнаружение активностей ферментов ACT и ЛДГ в крови больных. Динамика изменений этих активностей также представлена на этом рисунке. Активность ACT в норме составляет 5-40МЕ/л. При инфаркте миокарда активность ACT повышается через4-6ч; максимум активности наблюдают в течение 2-3дней. Уровень ЛДГ также увеличивается в плазме крови через несколько часов после закупорки кровеносного сосуда; максимум активности наблюдают на3-4-йдень, затей наступает постепенная нормализация активности. Уровень повышения активности ЛДГ коррелирует с размерами повреждения сердечной мышцы. 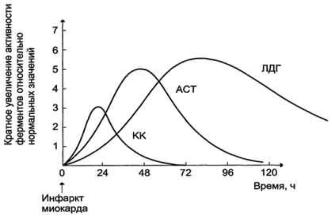
Аланинаминотрансфераза (АЛТ, АлАТ) – норма, результат при заболеваниях печени Нормальная активность АЛТ в крови мужчин равна 10-40Ед/л, у женщин –12-32Ед/л.Различные уровни
повышения активности АЛТ выявляются при острых гепатитах, циррозе печени, обтурационной желтухе и при приеме гепатотоксических препаратов (яды, некоторые антибиотики).
Резкое повышение активности АЛТ в 5-10и более раз является несомненным признаком острого заболевания печени. Причем такое повышение выявляется еще до того как проявятся клинические симптомы (желтуха, боли и прочее). Увеличение активности АЛТ можно засечьза1-4недели до проявления клиники и начать соответствующее лечение, не дав развиться заболеванию в полной мере. Высокая активность фермента при таком остром заболевании печени после проявления клинических симптомов держится недолго. Если нормализация активности ферментане происходит в течение двух недель, это говорит о развитии массивного повреждения печени.
|
Свёртывающая система крови. Этапы образования фибринового сгустка.
При повреждении кровеносного сосуда инициируется каскад реакций, в результате которого образуется сгусток крови - тромб, предотвращающий кровотечение. Основную роль в свёртывании (коагуляции) крови играют тромбоциты и ряд белков плазмы крови.
В остановке кровотечения различают 3 этапа. На первом этапе происходит сокращение кровеносного сосуда. Затем к месту повреждения прикрепляются тромбоциты, которые, наслаиваясь друг на друга, образуют тромбоцитарную пробку (белый тромб). Белый тромб является непрочным и может закупорить только небольшой кровеносный сосуд. На третьем этапе растворимый белок плазмы крови фибриноген превращается в нерастворимый белок фибрин, который откладывается между тромбоцитами, и формируется прочный фибриновый тромб. Такой тромб содержит эритроциты и поэтому называется красным тромбом. Образованию фибринового тромба предшествует каскад протеолитических реакций, приводящий к активации фермента тромбина, который и превращает фибриноген в фибрин. Все белки, участвующие в свёртывании крови, называют факторами свёртывания. Они синтезируются в основном в печени и клетках крови в виде неактивных предшественников.
Образование фибринового тромба начинается с превращения растворимого белка плазмы крови фибриногена в нерастворимый фибрин.
Фибриноген (фактор I) - гликопротеин Он синтезируется в печени и содержится в плазме крови в концентрации 8,02-12,9мкмоль/л (2 - 4 г/л). Молекула фибриногена состоит из шести полипептидных цепей, которые связаны друг с другом дисульфидными связями. Состав полипептидных цепей молекулы фибриногена обозначают Аα2, Вβ2, γ2
В образовании фибринового тромба можно выделить 4 этапа.
1.Превращение фибриногена в мономер фибрина. Сначала молекулы фибриногена освобождаются от отрицательно заряженных фрагментов А и В, в результате чего образуются мономеры фибрина. Превращение фибриногена (фактор I) в фибрин (фактор 1а) катализирует фермент тромбин (фактор Па). В каждой молекуле фибриногена тромбин гидролизует четыре пептидные связи аргинилглицил, две из которых соединяют фрагменты А с α-цепью, а две другие - В с β-цепьюв Аα2- иВβ2-цепяхфибриногена. Мономер фибрина, образующийся из фибриногена, имеет состав (α, β, γ)2.
2.Образование нерастворимого геля фибрина. На втором этапе образуется нерастворимый полимерный фибриновый сгусток - гель фибрина. В результате превращения фибриногена в фибрин-мономерв домене E открываются центры связывания с доменами D. Причём домен E содержит центры агрегации, формирующиеся только после частичного протеолиза фибриногена под действием тромбина, а домен D является носителем постоянных центров агрегации. Первичная агрегация молекул фибрина происходит в результате взаимодействия центров связывания домена E одной молекулы с комплементарными им участками на доменах D других молекул. После достижения протофибриллами определённой критической длины начинается их латеральная ассоциация, ведущая к образованию толстых фибриновых волокон.Образовавшийся гель фибрина непрочен, так как молекулы фибрина в нём связаны между собой нековалентными связями.
3.Стабилизация геля фибрина. В результате образования амидных связей между остатками лизина одной молекулы фибрина и остатками глутамина другой молекулы гель фибрина стабилизируется. Реакцию трансамидирования катализирует фермент трансглутамидаза (фактор ХIIIа).Фактор XIII активируется частичным протеолизом под действием тромбина.
4. Ретракция фибринового сгустка. Сжатие (ретракцию) геля обеспечивает актомиозин тромбоцитов - сократительный белок тромбостенин, обладающий АТФ-азнойактивностью. Тромбостенин участвует также в активации и агрегации тромбоцитов. Ретракция кровяного сгустка предупреждает полную закупорку сосудов, создавая возможность восстановления кровотока.
|
Внутренний и внешний пути свёртывания. Витамин К в свёртывании крови.
Витамин К и система свертывания крови
о внешнем пути свертывания кровиучаствуют тромбопластин (тканевой фактор, факторIII), проконвертин (факторVII), фактор Стюарта (факторX), проакцелерин (факторV), а также Са2+и фосфолипиды мембранных поверхностей, на которых образуется тромб. Гомогенаты многих тканей ускоряют свёртывание крови: это действие называют тромбопластиновой активностью. Вероятно, она связана с наличием в тканях какого-то специального белка. ФакторыVIIиX- проферменты. Они активируются путём частичного протеолиза, превращаясь в протеолитические ферменты - факторыVIIа иXа соответственно. ФакторV– это белок, который при действии тромбина превращается в факторV', который не является ферментом, но активирует ферментXа по аллостерическому механизму; активация усиливается в присутствии фосфолипидов и Са2+.
Свёртывание крови по внутреннему механизмупроисходит значительно медленнее и требует 10-15 мин. Этот механизм называют внутренним, потому что для него не требуется тромбопластин (тканевой фактор) и все необходимые факторы содержатся в крови. Внутренний механизм свёртывания также представляет собой каскад последовательных активаций проферментов. Начиная со стадии превращения фактораXвXа, внешний и внутренний пути одинаковы. Как и внешний путь, внутренний путь свёртывания имеет положительные обратные связи: тромбин катализирует превращение предшественниковVиVIIIв активаторыV' иVIII', которые в конечном итоге увеличивают скорость образования самого тромбина.
Внешний и внутренний механизмы свёртывания крови взаимодействуют между собой. Фактор VII, специфичный для внешнего пути свёртывания, может быть активирован факторомXIIа, который участвует во внутреннем пути свёртывания. Это превращает оба пути в единую систему свёртывания крови.
Биологическая роль витамина К обусловлена участием его в процессе свертывания крови. Он синтезирует в печени активные формы протромбина - вещества, обеспечивающего нормальное свертывание крови. свёртывание крови (протромбин (фактор II), факторы VII, IX, X, белок C, белок S и белок Z).
|
Противосвёртывающая система крови.
Физиологические ингибиторы свёртывания крови играют важную роль в поддержании гемостаза, так как они сохраняют кровь в жидком состоянии и препятствуют распространению тромба за пределы повреждённого участка сосуда.
Тромбин, образующийся в результате реакций прокоагулянтного и контактного путей свёртывания крови, вымывается током крови из тромба. Он может инактивироваться при взаимодействии с ингибиторами ферментов свёртывания крови или активировать антикоагулянтную фазу, тормозящую образование тромба.
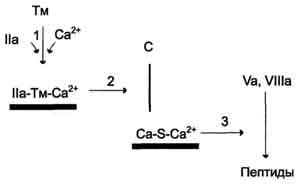
Антикоагулянтная фаза. Свёртывание крови должно быть ограничено не только в пространстве, но и во времени. Антикоагулянтная фаза ограничивает время существования активных факторов в крови и инициируется самим тромбином. Следовательно, тромбин, с одной стороны, ускоряет свёртывание крови, являясь последним ферментом каскада реакций коагуляции, а с другой - тормозит его, вызывая образование ферментных комплексов антикоагулянтной фазы на неповреждённом эндотелии сосудов. Этот этап представляет собой короткий каскад реакций, в котором кроме тромбина участвуют белок-активатортромбомодулин (Тм), витаминК-зависимаясериновая протеаза протеин С,белок-активаторS и факторы Va и VIIIa
Тромбомодулин - интегральный белок мембран эндотелиальных клеток. Он не требует протеолитической активации и служит белком-активаторомтромбина. Тромбин приобретает способность активировать протеин С только после взаимодействия с тромбомодулином, причём связанный с тромбомодулином тромбин не может превращать фибриноген в фибрин, не активирует фактор V и тромбоциты.
Протеин С - профермент, содержащий остатки γ-карбоксиглутамата.Тромбин в мембранном комплексе IIа-Тм-Са2+активирует частичным протеолизом протеин С. Активированный протеин С (Са) образует с белкомактиватором S мембраносвязанный
комплекс Ca-S-Са2+.Са в составе этого комплекса гидролизует в факторах Va и VIIIa по две пептидные связи и инактивирует эти факторы. Под действием комплексаCa-S-Са2+в течение 3 мин. теряется 80% активности факторов VIIIa и Va. Таким образом, тромбин по принципу положительной обратной связи не только ускоряет своё образование, но и, активируя протеин С, тормозит процесс свёртывания крови.
Наследственный дефицит протеина С и S ведёт к снижению скорости инактивации факторов VIIIa и Va и сопровождается тромботической болезнью. Мутация гена фактора V, при которой синтезируется фактор V, резистентный к протеину С, также приводит к тромбогенезу.
Антикоагулянтная фаза вызывает торможение каскада реакций свёртывания крови, а ингибиторы ферментов свёртывания инактивируют активные ферменты в кровяном русле.
Ингибиторы ферментов свёртывания крови. Физиологические ингибиторы ферментов свёртывания крови ограничивают распространение тромба местом повреждения сосуда. Белок плазмы крови антитромбин III - наиболее сильный ингибитор свёртывания крови; на его долю приходится около 80-90%антикоагулянтной активности крови. Он инактивирует ряд сериновых протеаз крови: тромбин, факторы IХа, Ха, ХIIа, калликреин, плазмин и урокиназу. Антитромбин III не ингибирует фактор VIIIa и не влияет на факторы в составе мембранных комплексов, а устраняет ферменты, находящиеся в плазме крови, препятствуя распространениютромбо-образованияв кровотоке.
Взаимодействие антитромбина с ферментами свёртывания крови ускоряется в присутствии гепарина. Гепарин - гетерополисахарид, который синтезируется в тучных клетках. В результате взаимодействия с гепарином антитромбин III приобретает конформацию, при которой повышается его сродство к сериновым протеазам крови. После образования комплекса антитромбин III-гепарин-ферментгепарин освобождается из него и может присоединяться к другим молекулам антитромбина.
При наследственном дефиците антитромбина III в молодом возрасте наблюдают тромбозы и эмболии сосудов, опасные для жизни.
α2-Макроглобулинобразует комплекс с сериновыми протеазами крови. В таком комплексе их активный центр полностью не блокируется, и они могут взаимодействовать с субстратами небольшого размера. Однако высокомолекулярные субстраты, например фибриноген, становятся недоступными для действия протеаз в комплексеα2-макроглобулинтромбин.
Антиконвер гин (тканевый ингибитор внешнего пути свёртывания) синтезируется в эндотелии сосудов. Он специфически соединяется с ферментным комплексом Тф-VIIа-Са2+,после чего улавливается печенью и разрушается в ней.
α1-Антитрипсинингибирует тромбин, фактор ХIа, калликреин, однако он не рассматривается как важный ингибитор факторов свёртывания крови,α1-Антитрипсинв основном на тканевом уровне ингибирует панкреатические и лейкоцитарные протеазы, коллагеназу, ренин, урокиназу.
Пептиды, образующиеся в результате протеолитической активации проферментов и профакторов, тоже обладают выраженными антикоагулянтными свойствами, но механизм их действия в настоящее время не выяснен
|
Нарушения коагуляционного гемостаза:гемофилии.
Наследственных коагулопатии доминируют (около 97%) гемофилии А и В
В основе обоих видов гемофилии лежит мутация локусов синтеза фактора VIII (гемофилия А) или фактора IX (гемофилия В) в Х-хромосоме.Болезнь при обеих формах наследуется по рецессивному, сцепленному с полом типу; носителями болезни являются женщины, а больными — только лица мужского пола (исключения из этого правила крайне редки). Все дочери больного гемофилией, получившие патологическуюХ-хромосомуот отца, являются кондукторами болезни, а сыновьяженщин-носительницгемофилии в 50% случаев имеют шанс стать больными, дочери (в 50% случаев) — носителями патологического гена.
Для гемофилии характерны кровоточивость гематомного типа — болезненные кровоизлияния в крупные суставы (гемартрозы), мышцы, забрюшинную клетчатку, в область черепа, гематурия, тяжелые отсроченные посттравматические и послеоперационные кровотечения, в том числе при малых травмах и вмешательствах (при порезах, удалении зубов и т. п.). Поскольку факторы VIII и IX участвуют только во внутреннем механизме свертывания крови, при гемофилии удлинены общее время свертывания цельной крови, время свертывания рекальцифицированной цитратной плазмы и активированного парциального тромбопластинового времени (АПТВ), тогда как протромбиновый показатель и тромбиновое время свертывания остаются нормальными.
К группе наследственных коагулопатий относят также болезнь Виллебранда, при которой тромбоцитопатия сочетается с дефицитом фактора VIII. В этом случае заболевание проявляется петехиальногематомной кровоточивостью в связи с сочетанным нарушением адгезивно-агрегационныхсвойств тромбоцитов и снижением коагуляционной активности крови. Это обусловлено тем, что фактор Виллебранда является переносчиком плазменного фактора VIII. При отсутствии фактора Виллебранда фактор VIII подвергается ускоренному разрушению в крови, что и обусловливает его дефицит и связанную с ним спонтанную гематомную кровоточивость.
|
|
. |
|
|
 Скачать 0.64 Mb.
Скачать 0.64 Mb.