Предмет наследственность и изменчивость в популяциях людей. Задача выявление, изучение, профилактика и лечение наследственных болезней, предотвращение вредного воздействия среды
 Скачать 4.41 Mb. Скачать 4.41 Mb.
|

|
Вопрос 30 Молекулярно – генетические методы исследований.  Вопрос 31 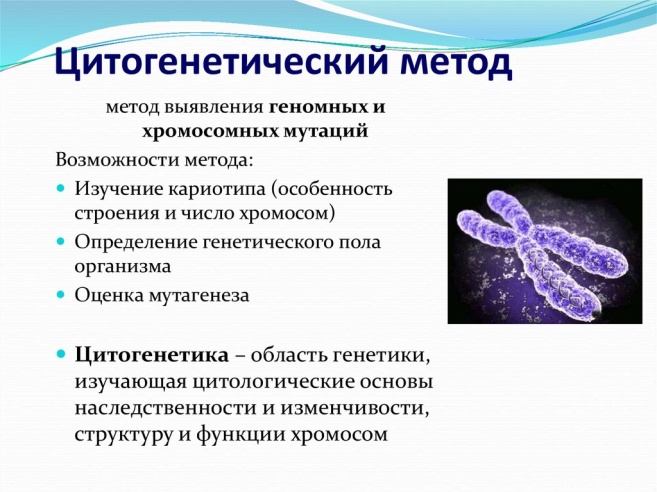 Метафазная хромосома состоит из двух продольных нитей ДНП – хроматид, соединенных друг с другом в области первичной перетяжки (центромеры). Центромера делит тело хромосомы на два плеча. В зависимости от расположения центромеры различают следующие типы хромосом: акроцентрические – центромера смещена к одному концу хромосомы и одно плечо очень короткое; субметацентрические – центромера смещена от середины хромосомы, и плечи имеют разную длину; метацентрические – центромера расположена посередине, и плечи примерно одинаковой длины. Участок каждого плеча вблизи центромеры называется – проксимальным, удаленный от нее –дистальным. Концевые отделы дистальных участков называются теломерами. Теломеры препятствуют соединению концевых участков хромосом. При потере этих участков наблюдаются хромосомные перестройки. Некоторые хромосомы могут иметь вторичные перетяжки, отделяющие от тел хромосомы участок, называемый спутником. Правила хромосом. Правило постоянства числа хромосом, Правило парности хромосом, Правило индивидуальности хромосом, Правило непрерывности хромосом. Метафазная хромосома состоит из двух продольных нитей ДНП – хроматид, соединенных друг с другом в области первичной перетяжки (центромеры). Центромера делит тело хромосомы на два плеча. В зависимости от расположения центромеры различают следующие типы хромосом: акроцентрические – центромера смещена к одному концу хромосомы и одно плечо очень короткое; субметацентрические – центромера смещена от середины хромосомы, и плечи имеют разную длину; метацентрические – центромера расположена посередине, и плечи примерно одинаковой длины. Участок каждого плеча вблизи центромеры называется – проксимальным, удаленный от нее –дистальным. Концевые отделы дистальных участков называются теломерами. Теломеры препятствуют соединению концевых участков хромосом. При потере этих участков наблюдаются хромосомные перестройки. Некоторые хромосомы могут иметь вторичные перетяжки, отделяющие от тел хромосомы участок, называемый спутником. Правила хромосом. Правило постоянства числа хромосом, Правило парности хромосом, Правило индивидуальности хромосом, Правило непрерывности хромосом.Денверская классификация хромосом, которая помимо размеров хромосом, учитывает их форму, положение центромеры и наличие вторичных перетяжек и спутников. 23 пары хромосом человека разбили на 7 групп от A до G. Важным параметром является центромерный индекс (ЦИ), который отражает отношение (в %) длины короткого плеча к длине всей хромосомы. К группе A относят 1-3 хромосомы. Это большие метацентрические и субметацентрические хромосомы, их центромерный индекс от 38-49. Группа B (4 и 5 пары). Это большие субметацентрические хромосомы, ЦИ 24-30. Группа C (6-12 пары). Хромосомы среднего размера, субметацентрические, ЦИ 27-35. К этой группе относят и Х-хромосому. Группа D (13-15 пары). Хромосомы акроцентрические, сильно отличаются от всех других хромосом человека, ЦИ около 15. Группа E (16-18 пары). Относительно короткие, метацентрические или субметацентрические, ЦИ 26-40. Группа F (19-20 пары): две короткие, субметацентрические хромосомы, ЦИ 36-46. Группа G (21-22 пары): это маленькие акроцентрические хромосомы, ЦИ 13-33. К этой группе относят и Y-хромосому. В основе Парижской классификации хромосом человека (1971 г.) лежат методы специальной дифференциальной их окраски, при которой каждой хромосоме выявляется характерный только для нее порядок чередования поперечных светлых и темных сегментов. Различные типы сегментов обозначают по методам, с помощью которых они выявляются наиболее отчетливо (Q-сегменты, G-сегменты, Т-сегменты, S-сегменты). Каждая хромосома человека содержит свойственную только ей последовательность полос, что позволяет идентифицировать каждую хромосому. Хромосомы спирализованы максимально в метафазе, менее спирализованы в профазе и прометафазе, что позволяет выделить большее число сегментов, чем в метафазе. На метафазной хромосоме приводятся символы, которыми принято обозначать короткое и длинное плечо, а также расположение районов и сегментов. В настоящее время существуют ДНК-маркеры или зонды, с помощью которых можно определить изменение определенного, даже очень маленького, сегмента в хромосомах (цитогенетические карты). На международном конгрессе генетики человека в Париже в 1971 г. (Парижская конференция по стандартизации и номенклатуре хромосом человека) была согласована система символов для более краткого и однозначного обозначения кариотипов. При описании кариотипа: • указывается общее число хромосом и набор половых хромосом, между ними ставится запятая (46, XX; 46, XY); • отмечается какая хромосома лишняя или какой не хватает (это указывается ее номером 5, 6 и др., или буквами данной группы А, В и др.); знаком «+» указывают на увеличение количества хромосом, знаком «-» указывают на отсутствие данной хромосомы 47, XY,+ 21; • плечо хромосомы, в котором произошло изменение (удлинение короткого плеча указывается символом (р+); укорочение (р-); удлинение длинного плеча указывается символом (q+); укорочение (q-); • символы перестроек (транслокация обозначается t, а делеция — del) помещают перед номерами вовлеченных хромосом, а перестроечные хромосомы заключают в скобки. Наличие двух структурно-аномальных хромосом обозначается точкой с запятой (;) или нормальной дробью (15/21). Идентификацию хромосом обеспечивает чередование темно и светло окрашенных полос. Вопрос 32   Вопрос 33 Медико-генетические аспекты брака. Медико-генетическое консультирование Существуют методы пренатальной диагностики. Как правило, пренатальный диагноз ставится в медико-генетических учреждениях. В России 7 центров федерального значения, 10 областных центров, 85 пунктов медико-генетического консультирования. Медико-генетическое консультирование — это один из видов специализированной помощи населению, направленной в первую очередь на предупреждение появления в семье детей с наследственной патологией. С этой целью составляют прогноз рождения в данной семье ребенка с наследственной болезнью, родителям объясняют вероятность этого события и оказывают помощь в принятии решения. В случае большой вероятности рождения больного ребенка родителям рекомендуют либо воздержаться от деторождения, либо провести пренатальную диагностику, если она возможна при данном виде патологии. Консультирование семей, обращающихся к врачу-генетику, включает три основных этапа. Как правило, за консультацией обращаются семьи, где уже имеется ребенок с наследственной патологией, или семьи, в которых имеются больные родственники. На первом этапе консультирования производится уточнение диагноза, что является необходимой предпосылкой любого консультирования. Уточнение диагноза в медико-генетической консультации проводят с помощью генетического анализа. Для этой цели используют генеалогический, цитогенетический, биохимический и другие требуемые методы исследований, которым подвергаются пробанд и его родственники. Точный клинический и генетический диагноз заболевания позволяет установить степень генетического риска и выбор эффективных методов пренатальной диагностики и профилактического лечения. На втором этапе консультирования делают прогноз потомства. Генетический риск может быть определен либо путем теоретических расчетов, основанных на генетических закономерностях, либо с помощью эмпирических данных. Сущность генетического прогноза заключается в определении вероятности появления наследственной патологии в семье. Наиболее эффективным является проспективное консультирование, когда риск рождения больного ребенка определяют до наступления беременности или в ранние ее сроки. Такие консультации чаще проводят в случае кровного родства супругов, при отягощенной наследственности по линии мужа или жены, при воздействии вредных средовых факторов на супругов незадолго до наступления беременности. Ретроспективное консультирование проводят после рождения больного ребенка относительно здоровья будущих детей. Определение прогноза потомства при разных формах наследственной патологии различно. При моногенных, менделирующих болезнях прогноз основывается на расчете вероятности появления потомства в соответствии с генетическими закономерностями. При этом, если известен тип наследования данного заболевания и по родословной удается установить генотип родителей, оценка риска сводится к анализу мевделевского расщепления. Если у пробанда установлена вновь возникшая мутация, то повторный риск рождения ребенка с такой же патологией незначителен. Расчет риска при моногенном заболевании может осложниться при пониженной экспрессивности или неполной пенетрантности гена, позднем проявлении генетической аномалии, генетической гетерогенности заболевания и вообще в случае неточного диагноза. При хромосомных болезнях определение риска повторного рождения потомства с хромосомными аномалиями зависит от того, нормальны ли кариотипы родителей, не обнаружено ли у них мозаицизма, не наблюдается ли семейной формы структурных аномалий хромосом. В случае отсутствия нарушений в кариотипе родителей вероятность повторного рождения второго ребенка с хромосомной аномалией оценивается по эмпирическим данным для каждого вида аномалии с учетом возраста родителей. При мультифакториальных заболеваниях, т. е. заболеваниях с наследственным предрасположением, основой оценки риска являются эмпирические данные о популяционной и семейной частоте каждого из них. Возможность проведения пренатальной диагностики является определяющей для принятия положительного решения в отношении завершения беременности. На третьем этапе консультирования врач-генетик в доступной форме объясняет семье степень генетического риска рождения наследственно аномального потомства, сущность пренатальной диагностики и помогает принять правильное решение в отношении деторождения. Однако окончательное решение этого вопроса остается за родителями. Широкое использование медико-генетического консультирования, разработка способов пренатальной диагностики наследственных заболеваний позволяют существенно уменьшить вероятность появления потомства с наследственной патологией в отдельных семьях. Чаще всего рецессивное потомство появляется у родителей с доминантным признаком, причем вероятность появления такого потомства возрастает в близкородственных браках, где оба родителя могут являться носителями одного и того же рецессивного аллеля, полученного от общего предка. Инбридинг приводит к повышению постоянства фенотипических признаков в потомстве и, в конечном итоге, производится для получения линий генетически идентичных особей (инбредные линии), на которых удобно проводить биологические и медицинские эксперименты. Как известно, организм получает каждый ген в двух экземплярах (аллелях) — от отца и от матери. Если эти гены различаются, то особь называется гетерозиготной (по данному гену), а если не различаются, то гомозиготной. При инбридинге родители являются родственниками и поэтому имеют много одинаковых генов, в результате чего гомозиготность увеличивается с каждым поколением. Первый этап консультирования начинается с уточнения диагноза болезни. В дальнейшем врач-генетик уточняет диагноз с помощью генетических анализов, использующих генеалогические, цитогенетические, специальные биохимические методы, разработанные именно для диагностики наследственных болезней. На втором этапе консультирования задача врача-генетика заключается в определении риска рождения больного ребенка. На третьем этапе консультирования врач-генетик выносит заключение о степени риска возникновения болезни у детей консультирующихся супругов и дает родителям соответствующие рекомендации. Заключительный этап консультирования — совет врача-генетика — не менее ответственный этап, требующий самого внимательного отношения. Вопрос 34   Вопрос 35 Общие подходы к лечению наследственных болезней сходны с подходами к лечению болезней любой другой этиологии. При лечении наследственных болезней полностью сохраняется принцип об индивидуализированном лечении - ведь врач и при наследственной патологии лечит не просто болезнь, а болезнь у конкретного человека. Возможно даже, что и при наследственной патологии принцип индивидуализированного лечения должен соблюдаться еще строже, потому что гетерогенность наследственных болезней далеко не расшифрована, а, следовательно, с одной и той же клинической картиной могут протекать разные наследственные болезни с различным патогенезом. В зависимости от условий пре - и постанального онтогенеза, а также от всего генотипа индивида фенотипические проявления мутаций у конкретного индивида могут модифицироваться в ту или другую сторону. Следовательно, необходима разная коррекция наследственной болезни у разных лиц. Как и при лечении других хорошо изученных болезней (например, инфекционных), можно выделить три подхода к лечению наследственных болезней и болезней с наследственной предрасположенностью: симптоматическое, патогенетическое, этиологическое. Применительно к наследственным болезням в отдельную группу можно выделить хирургические методы, поскольку они иногда выполняют функции симптоматической терапии, иногда патогенетической, иногда и той и другой вместе. При симптоматическом и патогенетическом подходах используются все методы современного лечения (лекарственное, генетическое, рентгенорадиологическое, физиотерапев-тическое, климатическое и т. д.). Генетический диагноз, клинические данные о состоянии больного и вся динамика болезни определяют поведение врача на протяжении всего периода лечения со строгим постоянным соблюдением гипполкратовского принципа "не навреди". При лечении наследственных болезней надо быть особенно внимательным в соблюдении этических и деонтологических принципов в отношении пациента и членов его семьи. Ведь часто речь идет о тяжелых хронических больных с детского возраста. Наследственные болезни настолько разнообразны по типам мутаций, по звеньям нарушенного обмена, степени вовлеченности в патологический процесс органов и систем, по характеру течения, что невозможно подробно описать лечение всех наследственных болезней. Изложим общие принципы лечения наследственной патологии и разработки новых методов. В целом можно ожидать дальнейших сдвигов в патогенетическом лечении путем возмещения продуктов (белков, гормонов) в связи с успехами физико-химической биологии, генной инженерии и биотехнологии: уже получают специфические белки и гормоны человека, необходимые для восполнения нарушенного звена обмена при лечении наследственных болезней (инсулин, соматотропин, интерферон). Развитие организма и поддержание гомеостаза зависят от согласованных взаимодействий множества генных продуктов, работающих в метаболических системах. Эти системы способны к адаптации, что обеспечивает гомеостаз в определенном диапазоне условий окружающей среды. Например, концентрацию глюкозы в крови регулируют продукты 30-40 генов; их взаимодействие обеспечивает относительно постоянный уровень глюкозы, несмотря на нерегулярное и крайне неравномерное потребление продуктов, являющихся ее источниками. Полагают, что для нормального развития и работы ЦНС необходимо взаимодействие примерно 10000 генных продуктов. Мутации, снижающие адаптивную способность подобных систем, вызывают патологические изменения, которые и называются наследственными болезнями. Иногда мутация так повреждает ту или иную систему, регулирующую развитие или гомеостаз, что она при любых обстоятельствах работает плохо либо не работает совсем, вызывая моногенную болезнь. В других случаях влияние мутантного гена в обычных условиях невелико, но некоторые факторы окружающей среды усугубляют его действие, вызывая пороки развития или нарушение гомеостаза ; это - полигенная болезнь. Таким образом, и в норме, и при патологии гены играют важную и неоднозначную роль. Поэтому и лечение наследственных болезней сложно и часто не вполне эффективно. Для лечения наследственных болезней необходимо: поставить точный диагноз; начать лечение до развития необратимых повреждений тканей; иметь четкое представление о патогенезе заболевания и о вызывающих его биохимических нарушениях. |