мр. Текст к лекции 2 Липиды (окончание) (1). Синтез жиров в жировой ткани и печени
 Скачать 0.59 Mb. Скачать 0.59 Mb.
|

|
Синтез ТАГ в печени. Образование ЛПОНП в печени и транспорт жиров в другие ткани Печень - основной орган, где идёт синтез жирных кислот из продуктов гликолиза. В гладком ЭР гепатоцитов жирные кислоты активируются и сразу же используются для синтеза жиров, взаимодействуя с глицерол 3-фосфатом. Как и в жировой ткани, синтез жиров идёт через образование фосфатидной кислоты. Синтезированные в печени жиры упаковываются в ЛПОНП и секретируются в кровь. В состав ЛПОНП, кроме жиров, входят холестерол, фосфолипиды и белок - апоВ-100. Это очень длинный белок - одна молекула апоВ-100 покрывает поверхность всего липопротеина. ОБМЕН ГЛИЦЕРОФОСФОЛИПИДОВ (синтез фосфатидилхолинов, фосфатидилэтаноламинов и фосфатидилсеринов) Начальные этапы синтеза глицерофосфолипидов и жиров происходят одинаково до образования фосфатидной кислоты. Фосфатидная кислота может синтезироваться двумя разными путями: через глицеральдегид-3-фосфат и через дигидроксиацетонфосфат.  Схема синтеза глицерофосфолипидов. R1 - радикал насыщенной жирной кислоты; R2 - радикал полиеновой жирной кислоты; SAM - S-аденозилметионин. 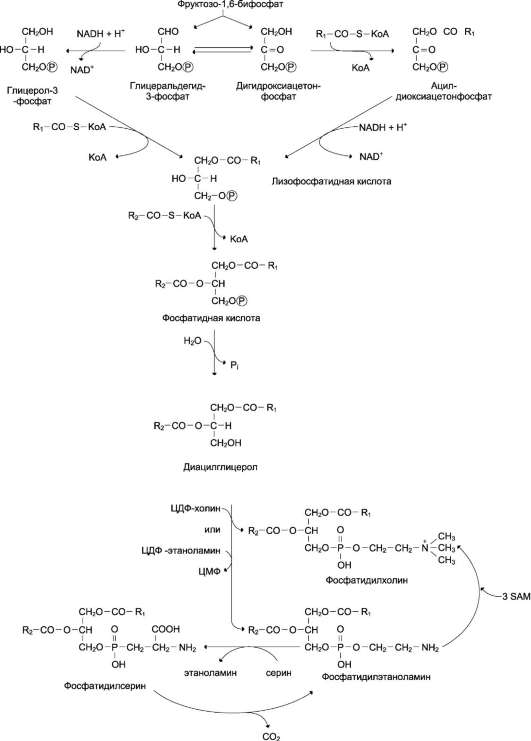 На следующем этапе фосфатидаза отщепляет от фосфатидной кислоты фосфатный остаток, в результате чего образуется диацилглицерол. Дальнейшие превращения диацилглицерола также могут идти разными путями. Один из вариантов - образование активной формы «полярной головки» фосфолипида: холин, серин или этаноламин превращаются в ЦДФ-холин, ЦДФ-серин или ЦДФ-этаноламин. Далее диацилглицерол взаимодействует c ЦДФ-производными, при этом выделяется ЦМФ, и образуется соответствующий фосфолипид, например фосфатидилхолин. Между глицерофосфолипидами возможны различные взаимопревращения. Фосфатидилхолин может образовываться и другим путём: из фосфатидилэтаноламина, получая последовательно 3 метильные группы от SAM. Фосфатидилсерин может превращаться в фосфатидилэтаноламин путём декарбоксилирования. Фосфатидилэтаноламин может превращаться в фосфатидилсерин путём обмена этаноламина на серин. Дипальмитоилфосфатидилхолин - основной компонент сурфактанта лёгких. Сурфактант - внеклеточный липидный слой с небольшим количеством гидрофобных белков, выстилающий поверхность лёгочных альвеол и предотвращающий слипание стенок альвеол во время выдоха. Основной компонент сурфактанта - дипальмитоилфосфатидилхолин, составляющий до 80% от всех фосфолипидов, входящих в состав сурфактанта. Кроме того, в сурфактант входят гидрофобные белки, общее количество которых не превышает 10-20%. Синтез дипальмитоилфосфатидилхолина (лецитина) в пневмоцитах II типа происходит в процессе эмбрионального развития и резко увеличивается в период от 32 до 36 нед. беременности. Важным показателем нормального формирования сурфактанта служит соотношение фосфатидилхолин/сфингомиелин >4. Это соотношение можно определять, исследуя состав амниотической жидкости. Недостаточное формирование сурфактанта у недоношенных детей после рождения приводит к развитию респираторного дистресс-синдрома - основной причины смерти у этой группы новорождённых. Соотношение фосфатидилхолин/сфингомиелин <2 указывает на высокий риск развития респираторного дистресс-синдрома. В случае необходимости лечение беременных кортикостероидами стимулирует синтез сурфактанта в лёгких плода и уменьшает риск развития респираторного дистресс-синдрома. Другой путь превращений диацилглицерола, при котором образуется активная форма - ЦДФ-диацилглицерол приводит к образованию фосфатидилинозитола и кардиолипина. Фосфатидилинозитол далее может фосфорилироваться с образованием фосфатидилинозитол-4,5-бисфосфата (инозитол-трифосфат), фосфолипида, располагающегося в наружной мембране клеток и участвующего в передаче гормональных сигналов внутрь клетки. Кардиолипин (дифосфатидилглицерол: два фосфатидилглицерола соединены с глицеролом) находится, главным образом, во внутренней мембране митохондрий и в небольшом количестве в сурфактанте лёгких. ХОЛЕСТЕРОЛ: ФУНКЦИИ, ОБМЕН Холестерол - стероид, характерный только для животных организмов. Он синтезируется во многих тканях человека, но основное место синтеза - печень. В печени синтезируется более 50% холестерола, в тонком кишечнике - 15-20%, остальной холестерол синтезируется в коже, коре надпочечников, половых железах. В сутки в организме синтезируется около 1 г холестерола; с пищей поступает 300-500 мг. 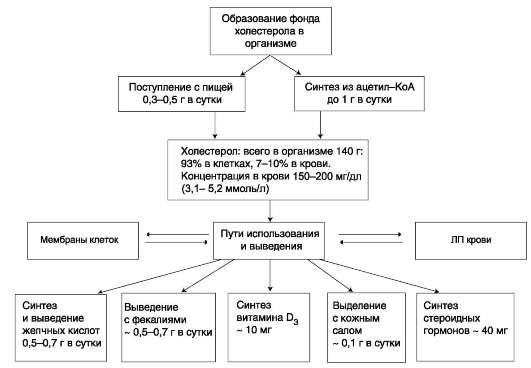 Фонд холестерола в организме, пути его использования и выведения. Холестерол выполняет много функций: входит в состав всех мембран клеток и влияет на их свойства, служит исходным субстратом в синтезе жёлчных кислот и стероидных гормонов. Предшественники в метаболическом пути синтеза холестерола превращаются также в убихинон - компонент дыхательной цепи и долихол, участвующий в синтезе гликопротеинов. Холестерол за счёт своей гидроксильной группы может образовывать эфиры с жирными кислотами. Этерифицированный холестерол преобладает в крови и запасается в небольших количествах в некоторых типах клеток, использующих его как субстрат для синтеза других веществ. Холестерол и его эфиры - гидрофобные молекулы, поэтому они транспортируются кровью только в составе разных типов ЛП. Обмен холестерола чрезвычайно сложен - только для его синтеза необходимо осуществление около 100 последовательных реакций. Всего в обмене холестерола участвует около 300 разных белков. Нарушения обмена холестерола приводят к одному из наиболее распространённых заболеваний - атеросклерозу. Смертность от последствий атеросклероза (инфаркт миокарда, инсульт) лидирует в общей структуре смертности населения. В развитии атеросклероза участвуют многие факторы, важнейшие из которых наследственные. Накопление холестерола в организме приводит к развитию и другого распространённого заболевания - желчнокаменной болезни. СИНТЕЗ ХОЛЕСТЕРОЛА И ЕГО РЕГУЛЯЦИЯ Реакции синтеза холестерола происходят в цитозоле клеток. Это один из самых длинных метаболических путей в организме человека. Сложный путь синтеза холестерола можно разделить на 3 этапа. I. Образование мевалоната Первый этап заканчивается образованием мевалоната (мевалоновой кислоты). Две молекулы ацетил-КоА конденсируются ферментом тиолазой с образованием ацетоацетил-КоА. Фермент гидроксиметилглутарил-КоА-синтаза присоединяет третий ацетильный остаток с образованием ГМГ-КоА (3-гидрокси-3-метилглутарил-КоА). Эта последовательность реакций сходна с начальными стадиями синтеза кетоновых тел. Однако реакции синтеза кетоновых тел происходят в митохондриях печени, а реакции синтеза холестерола - в цитозоле клеток. Следующая реакция, катализируемая ГМГ-КоА-редуктазой, является регуляторной в метаболическом пути синтеза холестерола. В этой реакции происходит восстановление ГМГ-КоА до мевалоната с использованием 2 молекул NADPH. Фермент ГМГ-КоА-редуктаза - гликопротеин, пронизывающий мембрану ЭР, активный центр которого выступает в цитозоль. 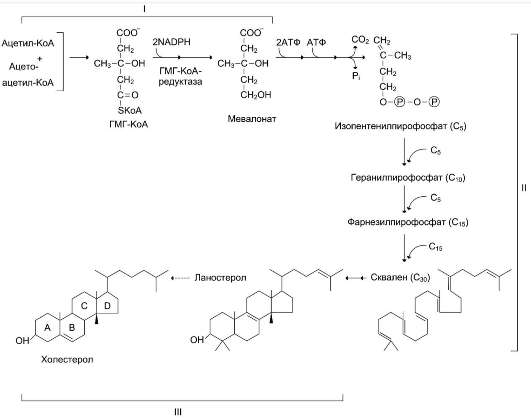 Синтез холестерола. С5 - изопентенилпирофосфат; С15 - фарнезилпирофосфат. Все атомы углерода холестерола происходят из ацетил-КоА. Сквален - углеводород линейной структуры - превращается ферментом циклазой в ланостерол, содержащий 4 конденсированных кольца и гидроксильную группу. Ланостерол через ряд последовательных реакций превращается в холестерол (I, II, III - этапы синтеза). II. Образование сквалена На втором этапе синтеза мевалонат превращается в пятиуглеродную изопреноидную структуру, содержащую пирофосфат - изопентенилпирофосфат. Продукт конденсации 2 изопреновых единиц - геранилпирофосфат. Присоединение ещё 1 изопреновой единицы приводит к образованию фарнезилпирофосфата - соединения, состоящего из 15 углеродных атомов. Две молекулы фарнезилпирофосфата конденсируются с образованием сквалена - углеводорода линейной структуры, состоящего из 30 углеродных атомов. III. Образование холестерола На третьем этапе синтеза холестерола сквален через стадию образования эпоксида ферментом циклазой превращается в молекулу ланостерола, содержащую 4 конденсированных цикла и 30 атомов углерода. Далее происходит 20 последовательных реакций, превращающих ланостерол в холестерол. На последних этапах синтеза от ланостерола отделяется 3 атома углерода, поэтому холестерол содержит 27 углеродных атомов. У холестерола имеется насыщенная разветвлённая боковая цепь из 8 углеродных атомов в положении 17, двойная связь в кольце В между атомами углерода в положениях 5 и 6, а также гидроксильная группа в положении 3. В организме человека изопентенилпирофосфат также служит предшественником убихинона (КоQ) и долихола, участвующего в синтезе гликопротеинов. В некоторых тканях гидроксильная группа холестерола этерифицируется с образованием более гидрофобных молекул - эфиров холестерола. Реакция катализируется внутриклеточным ферментом АХАТ (ацилКоА:холестеролацил-трансферазой). Эфиры холестерола - форма, в которой они депонируются в клетках или транспортируются кровью. В крови около 75% холестерола находится в виде эфиров. Регуляция синтеза холестерола Регуляция ключевого фермента синтеза холестерола (ГМГ-КоА-редуктазы) происходит разными способами. Фосфорилирование/дефосфорилирование ГМГ-КоА-редуктазы . При увеличении соотношения инсулин/глюкагон этот фермент дефосфорилируется и переходит в активное состояние. Действие инсулина осуществляется через 2 фермента: фосфатазу киназы ГМГ-КоА-редуктазы, которая превращает киназу в неактивное дефосфорилированное состояние; фосфатазу ГМГ-КоА-редуктазы путём превращения её в дефосфорилированное активное состояние. Результатом этих реакций служит образование дефосфорилированной активной формы ГМГ-КоА-редуктазы. Следовательно, в абсорбтивный период синтез холестерола увеличивается. В этот период увеличивается и доступность исходного субстрата для синтеза холестерола - ацетил-КоА (в результате приёма пищи, содержащей углеводы, так как ацетил-КоА образуется в основном при распаде глюкозы). В постабсорбтивном состоянии глюкагон через протеинкиназу А стимулирует фосфорилирование ГМГ-КоА-редуктазы, переводя её в неактивное состояние. Это действие усиливается тем, что одновременно глюкагон стимулирует фосфорилирование и инактивацию фосфатазы ГМГ-КоА-редуктазы и фосфорилирование киназы ГМГ-КоА-редуктазы, удерживая, таким образом, ГМГ-КоАредуктазу в фосфорилированном неактивном состоянии. В результате синтез холестерола в постабсорбтивном периоде и при голодании ингибируется. Ингибирование синтеза ГМГ-КоА-редуктазы. Конечный продукт метаболического пути (холестерол) снижает скорость транскрипции гена ГМГ-КоА-редуктазы, подавляя таким образом собственный синтез. В печени активно идёт синтез жёлчных кислот из холестерола, поэтому и жёлчные кислоты (как конечные продукты синтеза) подавляют активность гена ГМГ-КоА-редуктазы. Так как молекула ГМГ-КоА-редуктазы существует около 3 ч после синтеза, то ингибирование синтеза этого фермента конечным продуктом метаболического пути (холестеролом) является эффективной регуляцией. 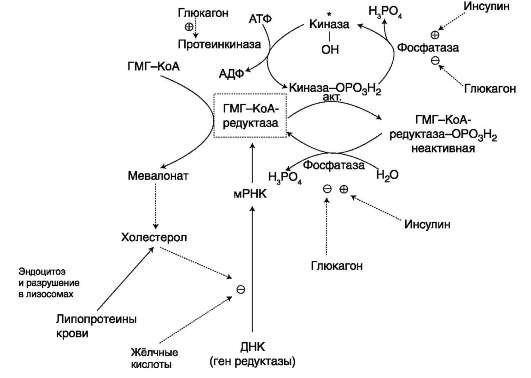 Регуляция активности ГМГ-КоА-редуктазы в печени. Холестерол и жёлчные кислоты снижают скорость транскрипции и, таким образом, синтез фермента. Инсулин стимулирует дефосфорилирование, а глюкагон - фосфорилирование ГМГ-КоА-редуктазы. Инсулин активирует 2 фосфатазы: киназы ГМГ-КоА-редуктазы* и фосфатазу, дефосфорилирующую непосредственно ГМГ-КоА-редуктазу. Глюкагон стимулирует фосфорилирование и инактивацию 2 фосфатаз и фосфорилирование и активацию киназы ГМГ-КоА-редуктазы. НАРУШЕНИЯ ЖИРОВОГО ОБМЕНА. ОЖИРЕНИЕ Жировая ткань составляет 20-25% от общей массы тела у женщин и 15-20% у мужчин. Однако избыточное накопление жира в адипоцитах (ожирение) широко распространено. Среди взрослого населения некоторых стран около 50% людей страдает ожирением. Ожирение - важнейший фактор риска развития инфаркта миокарда, инсульта, сахарного диабета, артериальной гипертензии и желчнокаменной болезни. Ожирением считают состояние, когда масса тела превышает 20% от «идеальной» для данного индивидуума. Образование адипоцитов происходит ещё во внутриутробном состоянии, начиная с последнего триместра беременности, и заканчивается в препубертатный период. После этого жировые клетки могут увеличиваться в размерах при ожирении или уменьшаться при похудании, но их количество существенно не изменяется в течение жизни, за исключением случаев гиперпластического ожирения. Первичное ожирение Первичное ожирение характеризуется множеством гормональных и метаболических особенностей у лиц, страдающих этим заболеванием. В самом общем виде можно сказать, что первичное ожирение развивается в результате алиментарного дисбаланса - избыточной калорийности питания по сравнению с расходами энергии. Суточные потребности организма в энергии складываются из: • основного обмена - энергии, необходимой для поддержания жизни; основной обмен измеряют по поглощению кислорода или выделению тепла человеком в состоянии покоя утром, после 12-часового перерыва в еде; • энергии, необходимой для физической активности. Затраты энергии, необходимые для физической активности, разделяют на 3 уровня: I - 30% энергии от основного обмена (у людей, ведущих сидячий образ жизни); II - 60-70% от энергии основного обмена (у людей, которые 2 ч в день имеют умеренную физическую нагрузку); III - 100% и более от энергии основного обмена (у людей, которые в течение нескольких часов в день занимаются тяжёлой физической работой). В зависимости от интенсивности нагрузки и возраста суточная потребность в энергии колеблется у женщин от 2000 до 3000 ккал в день, а у мужчин - от 2300 до 4000 ккал. Количество потребляемой пищи определяется многими факторами, в том числе и химическими регуляторами чувства голода и насыщения. Эти чувства определяются концентрацией в крови глюкозы и гормонов, которые инициируют чувство насыщения: холецистокинина, нейротензина, бомбезина, лептина. Причины первичного ожирения: • генетические нарушения (до 80% случаев ожирения - результат генетических нарушений); • состав и количество потребляемой пищи, метод питания в семье; • уровень физической активности; • психологические факторы. Метаболические различия между тучными и худыми людьми до настоящего времени не могут быть определены однозначно. Существует несколько теорий, объясняющих эти различия: • генетически детерминированная разница в функционировании «бесполезных» циклов (субстратных циклов). Эти циклы состоят из пары метаболитов, превращаемых друг в друга с помощью двух ферментов. Одна из этих реакций идёт с затратой АТФ. 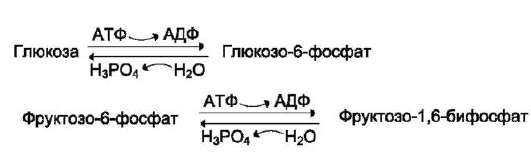 Например: • если эти субстраты превращаются друг в друга с одинаковой скоростью, то происходит «бесполезный» расход АТФ и, соответственно, источников энергии, например жиров; • у людей, склонных к ожирению, вероятно, имеется более прочное сопряжение дыхания и окислительного фосфорилирования, т.е. более эффективный метаболизм; • возможно, разное соотношение аэробного и анаэробного гликолиза. Анаэробный гликолиз (как менее эффективный) «сжигает» гораздо больше глюкозы, в результате снижается её переработка в жиры; • у отдельных индивидуумов имеется различие в активности Na+/К+-АТФазы, работа которой требует до 30% энергии, потребляемой клетками. Роль лептина в регуляции массы жировой ткани У человека и животных имеется «ген ожирения» - obese gene (ob). Продуктом экспрессии этого гена служит белок лептин, состоящий из 167 аминокислот, который синтезируется и секретируется адипоцитами и взаимодействует с рецепторами гипоталамуса. В результате его действия снижается секреция нейропептида Y. Нейропептид Y стимулирует пищевое поведение, поиск и потребление пищи у животных. Другие пептиды, участвующие в регуляции чувства сытости, например холецистокинин, также влияют на секрецию нейропептида Y. Таким опосредованным путём лептин выступает регулятором жировой массы, необходимой для роста и репродукции. Уровень лептина у больных ожирением может быть различным. У 80% больных концентрация лептина в крови тучных людей больше в 4 раза, чем у людей с нормальной массой тела. В этих случаях имеется генетический дефект рецепторов лептина в гипоталамусе, поэтому, несмотря на продукцию лептина, центр голода в гипоталамусе продолжает секрецию нейропептида Y. 20% больных имеют изменения в первичной структуре лептина. К настоящему времени описаны 5 одиночных мутаций в гене лептина, которые приводят к развитию ожирения. У этих больных наблюдают повышение отложения жиров в жировой ткани, чрезмерное потребление пищи, низкую физическую активность и развитие сахарного диабета типа II. Патогенез ожирения при дефекте гена ob может быть следующим: низкий уровень лептина в крови служит сигналом недостаточного количества запаса жиров в организме; этот сигнал включает механизмы, приводящие к увеличению аппетита и в результате к увеличению массы тела. Следовательно, можно сделать вывод о том, что первичное ожирение - не просто следствие переедания, а результат действия многих факторов, т.е. ожирение - полигенное заболевание. |