Госпитальная терапия. Перечень теоретических вопросов по дисциплине госпитальная терап. Вопросы дисциплины для студентов vi курса
 Скачать 1.99 Mb. Скачать 1.99 Mb.
|

|
Диагностика Общий анализ крови. Типичные для хронического миелоидного лейкоза изменения включают: анемию, присутствие единичных миелобластов и гранулоцитов на разной стадии дифференцировки; в период бластного криза количество бластных клеток увеличивается более чем на 20%. При хроническом лимфолейкозе определяющими гематологическими признаками выступают выраженный лейкоцитоз и лимфоцитоз, наличие лимфобластов и клеток Боткина-Гумпрехта. Пункции и биопсии. С целью определения морфологии опухолевого субстрата показано выполнение стернальной пункции, трепанобиопсии, биопсии лимфоузлов. В пунктате костного мозга при хроническом миелолейкозе увеличено количество миелокариоцитов за счет незрелых клеток гранулоцитарного ряда; в трепанобиоптате определяется замещение жировой ткани миелоидной. При хроническом лимфоидном лейкозе миелограмма характеризуется резким усилением лимфоцитарной метаплазии. Инструментальные исследования. Для оценки выраженности лимфопролиферативного синдрома применяются УЗИ лимфатических узлов, селезенки, рентгенография грудной клетки, лимфосцинтиграфия, МСКТ брюшной полости и ряд других. Лечение хронического лейкоза На ранней доклинической стадии лечение неэффективно, поэтому больные подлежат динамическому наблюдению. Общережимные мероприятия предполагают исключение физических перегрузок, стрессов, инсоляции, электропроцедур и теплолечения; полноценное витаминизированное питание, длительные прогулки на свежем воздухе. В развернутом периоде миелолейкоза назначается химиотерапевтическое лечение (бусульфан, митобронитол, гидроксимочевина и др.), при выраженной спленомегалии проводится облучение селезенки. Подобная тактика, хоть и не приводит к полному излечению, но существенно тормозит прогрессирование болезни и позволяет отсрочить наступление бластного криза. Кроме медикаментозной терапии, при хроническом миелоцитарном лейкозе используются процедуры лейкафереза. В ряде случаев излечение достигается с помощью трансплантации костного мозга. При переходе хронического миелолейкоза в терминальную стадию назначается высокодозная полихимиотерапия. В среднем после установления диагноза больные хроническим миелолейкозом живут 3-5 лет, в отдельных случаях – 10-15 лет. Также проводится цитостатическая терапия (хлорбутин, циклофосфамид), иногда в сочетании со стероидной терапией, облучением лимфоузлов, селезенки, кожи. При значительном увеличении селезенки выполняется спленэктомия. Применяется трансплантация стволовых клеток. 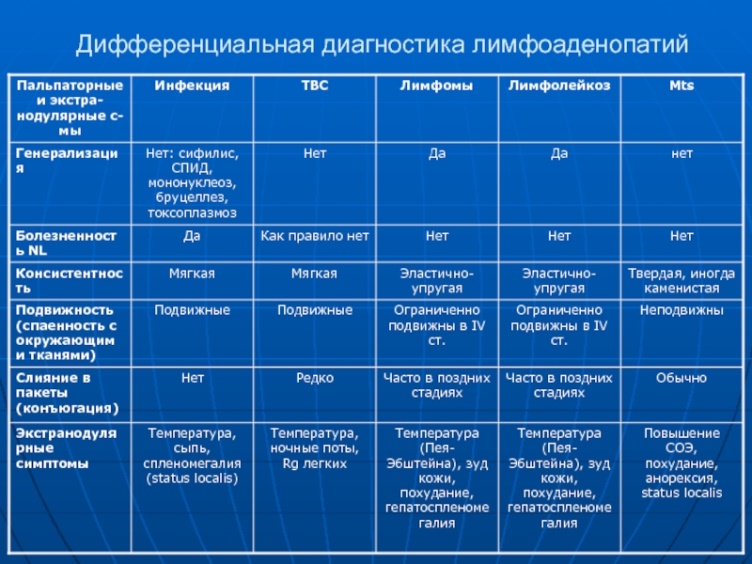 Эритремия: этиология, патогенез, клиника, диагностика. Дифференциальная диагностика при плеторическом синдроме. Исход, лечение. Эритремия (истинная полицитемия) – это Ph-негативное хроническое миелопролиферативное клональное заболевание, которое возникает на уровне стволовой кроветворной клетки и сопровождается чрезмерной пролиферацией эритроидного, миелоидного и мегакариоцитарного ростков в костном мозге (панмиелоз), что сопровождается эритроцитозом, высоким уровнем гемоглобина, лейкоцитозом, тромбоцитозом в крови (панцитоз), нередко с возникновением спленомегалии. Этиопатогенез Непосредственные причины заболевания до настоящего времени не изучены. Основным предположением является многоэтапность возникновения заболевания, где предрасположенность к болезни реализуется под воздействием внешних факторов, которые обладают способностью повреждать геном здоровой клетки, что становится причиной ее злокачественной трансформации. У преобладающего большинства клеток были выявлены активирующие мутации в гене JAK2. Из-за подобной мутации возникает гиперплазия не только эритроидной, но и гранулоцитарной и мегакариоцитарной линий дифференцировки с развитием так называемого «панмиелоза». Определяющим свойством гемопоэза при истинной полицитемии является способность клеток-предшественниц гемопоэза формировать эритроидные колонии в отсутствие экзогенного эритропоэтина. При первичной верификации диагноза цитогенетические изменения выявляют у 20 % больных. На поздних стадиях болезни частота хромосомных изменений достигает 80–100 %. Помимо активирующих мутаций в гене JAK2 у больных истинной полицитемией обнаруживаются мутации и в других генах. Клональный гемопоэз, фенотипические особенности истинной полицитемии и прогрессирование заболевания возникают вследствие приобретенных соматических генных мутаций, которые принимают участие в регуляции цитокиновых реакций, сплайсинге РНК, а также эпигенетической регуляции. Клиническая картина В течении истинной полицитемии выделяют три фазы: преполицитемическую – с пограничным или умеренным эритроцитозом; собственно полицитемическую – развернутая картина заболевания с выраженным эритроцитозом; постполицитемический миелофиброз с цитопениями, включая анемию, неэффективный гемопоэз, фиброз костного мозга, с экстрамедуллярным кровотечением и гиперспленизмом. Специфическая симптоматика истинной полицитемии отсутствует, и различные ее проявления можно выявить у преобладающего числа больных. Наиболее распространены жалобы на усталость, кожный зуд, ночную потливость, боль в костях и суставах, лихорадку и снижение массы тела. Кожный зуд – это особое неприятное субъективное ощущение, которое пациенты характеризуют как генерализованное чувство жжения, покалывания, пощипывания возникающее после контакта с водой (аквагенный зуд). Перепад температуры, употребление алкоголя или физические упражнения могут вызывать усиление симптоматики, которые могут продолжаться около 40 мин и нередко сопровождаются агрессией, раздражительностью, депрессией и суицидальными мыслями. Помимо того, у 35–45 % пациентов выявляется спленомегалия, которая является одним из проявлений прогрессирования заболевания. Спленомегалия приводит к появлению вторичных симптомов, таких как боль в животе, ощущение быстрого насыщения, потеря массы тела, тошнота. Увеличение селезенки может стать причиной компрессии органов брюшной полости, портальной гипертензии. Симптоматика опухолевой интоксикации, причиной которой является истинная полицитемия, и спленомегалия выявляются у 70 % больных и приводят к снижению качества жизни. Инструментом для полноформатной оценки симптомов у больных истинной полицитемией, которые позволяют провести оценку выраженность симптоматики интоксикации, являются опросники European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC-Qol) и MPN-Symptom Assessment Form (MPN-SAF). NB! Плеторический синдром (от слова «плетора» – полнокровие) – характеризуется увеличением массы циркулирующих эритроцитов, что приводит к появлению жалоб на головокружение, головные боли, ухудшение зрения, кожный зуд после мытья, жгучие боли и парестезии в кончиках пальцев, приступы стенокардии. При осмотре кожа и видимые слизистые оболочки с синюшним оттенком (положительный симптом Купермана). Сосудистые осложнения – тромбозы любой локализации, приступы покраснения пальцев рук и ног, которые сопровождаются болью и жжением (эритромелалгия). Увеличение объёма циркулирующих эритроцитов приводит к появлению артериальной гипертензии у пациентов, которые до начала заболевания не жаловались на данный симптом, либо усугубление уже имеющейся гипертонической болезни, которая плохо поддаётся лечению традиционными гипотензивными препаратами. Более выраженными становятся симптомы ишемической болезни сердца, церебрального атеросклероза. В раннюю стадию заболевания отмечается эритроцитоз, в костном мозге – панмиелоз. Так как заболевание развивается постепенно, от его начала до постановки диагноза проходит от 2 до 4 лет. Длительность данной стали – до 5 лет. NB! Миелопролиферативный синдром обусловлен гиперплазией трех ростков кроветворения. Проявляется в виде кожного зуда, потливости, слабости, повышенной температуры тела, болей в костях. Повышенный распад гранулоцитов сопровождается нарушением уратового обмена, что проявляется в виде мочекислого диатеза, камнеобразования в почках, подагры, подагрической полиартралгии. Спленомегалия может быть обусловлена увеличением секвестрирующей функции селезенки. Диагностика и дифференциальный диагноз Верификация диагноза истинной полицитемии проводится на основании стандартных классификационных критериев, утвержденных ВОЗ в 2008 г.: Большие критерии: Гемоглобин более 185 г/л у мужчин и 165 г/л женщин или другие признаки повышения объема циркулирующих эритроцитов. Гематокрит более 52 % у мужчин и более 48 % у женщин. Мутация гена JAK2V617F или в экзоне 12. Малые критерии: По результатам биопсии костного мозга – трехростковая гиперплазия (панмиелоз): усиление пролиферативной активности элементов эритроидного, гранулоцитарного, мегакариоцитарного ростков миелопоэза. Уровень эритропоэтина (ЭПО) сыворотки ниже референсных показателей. Формирование эндогенных эритроидных колоний в культуре костномозговых клеток больного без добавления эритропоэтина. Диагноз считается верифицированным в случае выявления одного или двух больших критериев и одного малого или одного большого и двух малых критериев. Критерии диагноза постполицитемического миелофиброза Необходимые критерии: Наличие ранее документированного на основании критериев ВОЗ диагноза «истинная полицитемия». Фиброз костного мозга 2–3-й или 3–4-й степени. Дополнительные критерии (не менее двух): Анемия или длительное отсутствие потребности в кровопусканиях или в циторедуктивной терапии для коррекции эритроцитоза. Лейкоэритробластическая картина периферической крови (наличие незрелых гранулоцитов или эритробластов). Спленомегалия (> 5 см из-под края реберной дуги). Наличие больше одного из трех конституциональных симптомов: потеря более 10 % веса за 6 месяцев, ночные поты, необъяснимая лихорадка (> 37,5º C). Лечение Подход к лечению больных истинной полицитемией основан на оценке факторов риска тромбоэмболических осложнений. Всем пациентам рекомендуется отказ от табакокурения, нормализация артериального давления, устранение гиперхолестеринемии и гиперглюкоземии, нормализация веса. Проводится посиндромная симптоматическая и патогенетическая терапия. Пациенты могут быть стратифицированы в группы высокого или низкого риска на основании возраста и тромбозов в анамнезе. Категория «промежуточный риск» включает в себя молодых пациентов с сердечно-сосудистыми факторами риска при отсутствии тромбозов в анамнезе. Пациенты группы низкого риска подлежат активному динамическому наблюдению. С целью контроля гематокрита больным проводят сеансы кровопусканий/эритроцитафереза. Для профилактики тромботических осложнений пациенты получают терапию антиагрегантами. Циторедуктивная терапия данной группе больных проводится в случае плохой переносимости кровопусканий, при прогрессировании заболевания, тромбоцитозе более 1500×109/л или нарастании лейкоцитоза. Больные, отнесенные к группе высокого риска, нуждаются в проведении циторедуктивной терапии. Для больных группы промежуточного риска вырабатывается индивидуальная тактика лечения. Рекомендации по лечению истинной полицитемии (Рабочая группа по изучению истинной полицитемии): Большинство больных со впервые диагностированной истинной полицитемией должны получать лечение кровопусканиями с целью достижения уровня гематокрита не более 0,45, с последующим динамическим наблюдением за развитием симптоматики заболевания. При индивидуализации терапии обязательно учитывается возраст пациентов. Лечение больных моложе 50-ти лет, без тромботических осложнений в анамнезе и тяжелого гипертромбоцитоза (< 1000×109/л) может предусматривать только кровопускания в сочетании с терапией аспирином (или без него) в дозе 100–375 мг в сутки. Пациентам старше 70-ти лет с тромботическими осложнениями в анамнезе и тяжелым гипертромбоцитозом рекомендуется проведение терапии миелосупрессивными фармсредствами. Больные 50–70 лет без тромботических осложнений и тяжелого гипертромбоцитоза получают лечение миелосупрессивными агентами или кровопусканиями, хотя при последнем виде лечения может увеличиваться риск тромботических осложнений. Возможно привлечение пациентов для включения в программу клинических исследований по использованию таргетной терапии. Фармсредством выбора для проведения миелосупрессии является препарат гидроксимочевины. В случаях гипертромбоцитоза возможно использование интерферона-альфа. Прогноз у пациентов с истинной полицитемией достаточно благоприятный: продолжительность жизни может составлять 10–20 лет (в среднем 13,5 лет), нередко превышает 20 лет. Парапротеинемические гемобластозы. Основные формы, диагностика. Клинические проявления. Дифференциальная диагностика парапротеинемий. Лечение. Парапротеинемические гемобластозы — особая группа опухолей лимфатической системы, при которых опухолевые клетки (лимфоциты или плазматические) синтезируют иммуноглобулин. К парапротеинемическим гемобластозам относятся миеломная болезнь, макроглобулинемия Вальденстрема, болезнь тяжелых цепей. Эти лейкозы своим клеточным субстратом имеют преимущественно зрелые элементы - плазмоциты, лимфоциты, поэтому должны относиться к группе хронических лейкозов. Клиническая картина в начале болезни не имеет типичных черт. По мере прогрессирования процесса возникают боль в позвонках, корешковый синдром, боль в ребрах, обусловленные разрушением (гнездным или диффузным) костей растущей плазмоцитомой. Иногда ведущими становятся признаки, связанные с угнетением продукции костного мозга: анемия, гранулоцитопения с ее инфекционными осложнениями, тромбоцитопенический геморрагический синдром. В других случаях ведущим симптомом довольно быстро становится нарастающая протеинурия, почечная недостаточность. Диагноз устанавливают на основании обнаружения высокого содержания плазматических (иногда атипичных) клеток в костном мозге (выше 15%), появления моноклонового иммуноглобулина в сыворотке крови (нередко и в моче) в виде узкой полосы на электрофореграмме (М-градиент), снижения уровня нормальных иммуноглобулинов (поданным иммуноэлектрофореза). В сомнительных случаях, например при несекретирующей миеломе, отсутствии высокого процента плазмоцитов в костном мозге, диагноз устанавливают в специализированном учреждении. Подозрительны следующие признаки: немотивированное нарастающее на протяжении ряда лет повышение СОЭ (следствие избытка гамма-глобулинов в крови), упорная протеинурия (не альбуминурия, а глобулинурия, устанавливаемая методом электрофореза мочи), проявление остеодеструктивных очагов, анемия, лейкоците- и тромбоцитопения. Лечение обычно начинают в стационаре, назначая цитостатики - циклофосфан или сарколизин вместе с преднизолоном и метандростенолоном (неробол) или ретаболилом. Сарколизин назначают по 10 мг/сут через день или ежедневно, по 200-300 мг на курс. В течение всего курса дают преднизолон - 10-20 мг/сут, ретаболил по 50 мг в/в 1 раз в неделю (или неробол по 10-15 мг ежедневно). После завершения курса переходят к поддерживающей терапии сарколизином по 10 мг 1 раз в 5-10 дней. При почечной недостаточности сарколизин становится крайне опасным, так как он выводится почками и нарушение его выведения ведет к резкому повышению концентрации цитостатика в крови с развитием глубокой панцитопении. Вместо сарколизина можно использовать циклофосфан по 400 мг через день внутрь, в/в или в/м (на курс 8-10 г). В течение всего курса преднизолон и неробол (или ретаболил) дают в той же дозе, что и в курсе с сарколизином. После окончания курса проводят поддерживающую терапию (по 400 мг циклофосфана в день 1 раз в 5-10 дней). Существуют и другие схемы лечения миеломной болезни, требующие участия специалиста. При снижении уровня лейкоцитов до 1000 в 1 мкл необходима отмена цитостатика. Локальные очаги поражения часто требуют лучевой терапии. Дифференциальный диагноз следует проводить с плазмоклеточным лейкозом, отличающимся от множественной миеломы значительно более быстрым прогрессированием, наличием значительного количества плазматических клеток в крови, тромбоцитопенией с выраженным геморрагическим синдромом, обширной плазматической инфильтрацией различных органов. Необходима дифференциальная диагностика от других заболеваний, сопровождающихся гипергаммаглобулинемией. В первую очередь, от макроглобулинемии Вальденстрема, при которой имеет место избыточное содержание в крови иммуноглобулинов-макроглобулинов класса IgM. В отличие от ММ при этом заболевании имеет место выраженная лимфаденопатия, спленомегалия, гепатомегалия. Возможно формирования криоглобулинемического васкулита, синдрома Рейно, холодовой иммунной гемолитической анемии. Не возникает поражения почек. Отсутствует остеолиз черепа, позвонков, других костей скелета. Геморрагические диатезы: классификация, типы кровоточивости, этиология, патогенез клинические проявления, диагностика, лечение тробоцитопенической пурпуры. Дифференциальная диагностика при геморрагическом синдроме. Геморрагические диатезы (ГД) - группа врожденных или приобретенных болезней и синдромов, основным клиническим признаком которых служит повышенная кровоточивость - склонность к повторным кровотечениям или кровоизлияниям, возникающим спонтанно или после незначительных травм. КЛАССИФИКАЦИЯ 1. Тромбоцитопении (снижение количества тромбоцитов) и тромбоцитопатии (нарушение функциональных свойств тромбоцитов). 2. Коагулопатии (гемофилии), развивающиеся: - при недостаточном содержании прокоагулянтов, участвующих в плазменном звене гемостаза; - недостаточной функциональной активности прокоагулянтов; - присутствии в крови ингибиторов отдельных прокоагулянтов. 3. Ангиопатии (вазопатии) - повреждение сосудистой стенки врожденной этиологии или развивающееся в результате иммуноаллергического или инфекционно-токсического воздействия. 4. Избыточный фибринолиз, возникающий: - при лечении тромболитическими препаратами; - дефекте ингибитора плазмина или избытке тканевого активатора плазминогена наследственной этиологии. 5. ДВС-синдром, представленный сочетанием нарушений различных компонентов гемостаза (тромбоцитопения, коагулопатия и др.). |