Госпитальная терапия. Перечень теоретических вопросов по дисциплине госпитальная терап. Вопросы дисциплины для студентов vi курса
 Скачать 1.99 Mb. Скачать 1.99 Mb.
|

|
ТИПЫ КРОВОТОЧИВОСТИ 1. гематомный (массивные, глубокие, напряженные и весьма болезненные кровоизлияния в крупные суставы, мышцы, подкожную жировую и забрюшинную клетчатку), этот тип характерен для гемофилий; 2. пятнисто-петехиальный (микроциркуляторный тип, характеризующийся безболезненными не напряженными, не сдавливающими окружающие ткани поверхностными кровоизлияниями в кожу и слизистые оболочки, петехиями, синяками, десневыми, носовыми и маточными кровотечениями) встречается при тромбоцитопениях и тромбоцитопатиях; 3. смешанный микроциркуляторно-гематомный (в клинической картине преобладает петехиально-пятнистая кровоточивость, гематомы немногочисленны, но достигают очень больших размеров и располагаются преимущественно в подкожной жировой или забрюшинной клетчатке) встречается при ДВСсиндроме и передозировке антикоагулянтов; 4. васкулитно-пурпурный (геморрагические высыпания на коже конечностей и туловища, как правило, симметричные и несколько приподняты вследствие воспалительной инфильтрации и отека). Отмечают при геморрагическом васкулите; 5. ангиоматозный (характеризуется отсутствием спонтанных и посттравматических кровоизлияний в кожу, подкожную жировую клетчатку и другие ткани и органы, но возникают весьма упорные кровотечения одной-двух локализаций, например, носовые, реже - гематурия, легочные и желудочно-кишечные). Этот тип кровоточивости отмечают при различных формах телеангиэктазии. Причины геморрагических диатезов Различают наследственные (первичные) геморрагические диатезы, манифестирующие в детском возрасте, и приобретенные, чаще всего являющиеся вторичными (симптоматическими). Первичные формы являются семейно-наследственными и связаны с врожденным дефектом или дефицитом обычно одного фактора свертывания. Примерами наследственных геморрагических диатезов служат гемофилия, тромбастения Гланцмана, болезнь Рандю-Ослера, болезнь Стюарта Прауэр и др. Исключение составляет болезнь Виллебранда, являющаяся полифакторной коагулопатией, обусловленной нарушением фактора VIII, сосудистого фактора и адгезивности тромбоцитов. К развитию симптоматических геморрагических диатезов обычно приводит недостаточность сразу нескольких факторов гемостаза. При этом может отмечаться уменьшение их синтеза, повышение расходования, изменение свойств, повреждение эндотелия сосудов и пр. Причинами повышенной кровоточивости могут служить различные заболевания (СКВ, цирроз печени, инфекционный эндокардит), геморрагические лихорадки (лихорадка денге, Марбург, Эбола, Крымская, Омская и др.), дефицит витаминов (С, К и др.). В группу ятрогенных причин входит длительная или неадекватная по дозе терапия антикоагулянтами и тромболитиками. Чаще всего приобретенные геморрагические диатезы протекают в форме синдрома диссеминированного внутрисосудистого свертывания (тромбогеморрагического синдрома), осложняющего самые различные патологии. Возможно вторичное развитие аутоиммунных, неонатальных, посттрансфузионных тромбоцитопений, геморрагического васкулита, тромбоцитопенической пурпуры, геморрагического синдрома при лучевой болезни, лейкозах и т. д. Симптомы геморрагических диатезов В клинике различных форм гемостазиопатий доминируют геморрагический и анемический синдромы. Выраженность их проявлений зависит от патогенетической формы геморрагического диатеза и сопутствующих нарушений. При различных видах геморрагических диатезов могут развиваться разные типы кровотечений. Микроциркуляторный (капиллярный) тип кровоточивости встречается при тромбоцитопатиях и тромбоцитопениях. Проявляется петехиально-пятнистыми высыпаниями и синяками на коже, кровоизлияниями в слизистые оболочки, кровотечениями после экстракции зуба, десневыми, маточными, носовыми кровотечениями. Геморрагии могут возникать при незначительном травмировании капилляров (при надавливании на кожу, измерении АД и пр.). Гематомный тип кровоточивости характерен для гемофилии, возможен при передозировке антикоагулянтов. Характеризуется образованием глубоких и болезненных гематом в мягких тканях, гемартрозов, кровоизлияний в подкожно-жировую и забрюшинную клетчатку. Массивные гематомы приводят к расслоению тканей и развитию деструктивных осложнений: контрактур, деформирующих артрозов, патологических переломов. По происхождению такие кровотечения могут быть спонтанными, посттравматическими, послеоперационными. Капиллярно-гематомные (смешанные) геморрагии сопровождают течение ДВС-синдрома, болезни Виллебранда, наблюдаются при превышении дозы антикоагулянтов. Сочетают петехиально-пятнистые кровоизлияния и гематомы мягких тканей. Микроангиоматозный тип кровоточивости встречается при геморрагическом ангиоматозе, симптоматических капилляропатиях. При этих геморрагических диатезах возникают упорные рецидивирующие кровотечения одной или двух локализации (обычно носовые, иногда - желудочно-кишечные, легочные, гематурия). Васкулитно-пурпурный тип кровоточивости отмечается при геморрагических васкулитах. Представляет собой мелкоточечные геморрагии, как правило, имеющие симметричное расположение на конечностях и туловище. После исчезновения кровоизлияний на коже длительно сохраняется остаточная пигментация. Частые кровотечения вызывают развитие железодефицитной анемии. Для анемического синдрома, сопровождающего течение геморрагических диатезов, характерны слабость, бледность кожных покровов, артериальная гипотония, головокружения, тахикардия. При некоторых геморрагических диатезах может развиваться суставной синдром (припухлость сустава, артралгии), абдоминальный синдром (тошнота, схваткообразные боли), почечный синдром (гематурия, боли в пояснице, дизурия). Диагностика Целью диагностики геморрагических диатезов служит определение его формы, причин и степени выраженности патологических сдвигов. План обследования пациента с синдромом повышенной кровоточивости составляется гематологом совместно с лечащим специалистом (ревматологом, хирургом, акушером-гинекологом, травматологом, инфекционистом и др.). В первую очередь исследуются клинические анализы крови и мочи, количество тромбоцитов, коагулограмма, кал на скрытую кровь. В зависимости от полученных результатов и предполагаемого диагноза назначается расширенная лабораторная и инструментальная диагностика (биохимическое исследование крови, стернальная пункция, трепанобиопсия). При геморрагических диатезах, имеющих иммунный генез, показано определение антиэритроцитарных антител (тест Кумбса), антитромбоцитарных антител, волчаночного антикоагулянта и др. Дополнительные методы могут включать функциональные пробы на ломкость капилляров (пробы жгута, щипка, манжеточную пробу и др.), УЗИ почек, УЗИ печени; рентгенографию суставов и др. Для подтверждения наследственной природы геморрагических диатезов рекомендуется консультация генетика. Лечение геморрагических диатезов При подборе лечения практикуется дифференцированный подход, учитывающий патогенетическую форму геморрагического диатеза. Так, при повышенной кровоточивости, вызванной передозировкой антикоагулянтов и тромболитиков, показана отмена данных препаратов или коррекция их дозы; назначение препаратов витамина К (викасола), аминокапроновой кислоты; переливание плазмы. Терапия аутоиммунных геморрагических диатезов основана на применении глюкокортикоидов, иммунодепрессантов, проведении плазмафереза; при нестабильном эффекте от их применения требуется проведение спленэктомии. При наследственном дефиците того или иного фактора свертываемости показано проведение заместительной терапии их концентратами, трансфузий свежезамороженной плазмы, эритроцитарной массы, гемостатической терапии. С целью местной остановки небольших кровотечений практикуется наложение жгута, давящей повязки, гемостатической губки, льда; проведение тампонады носа и пр. При гемартрозах выполняются лечебные пункции суставов; при гематомах мягких тканей – их дренирование и удаление скопившейся крови. Основные принципы лечения ДВС-синдрома включают активное устранение причины данного состояния; прекращение внутрисосудистого свертывания, подавление гиперфибринолиза, проведение заместительной гемокомпонентной терапии и т. д. 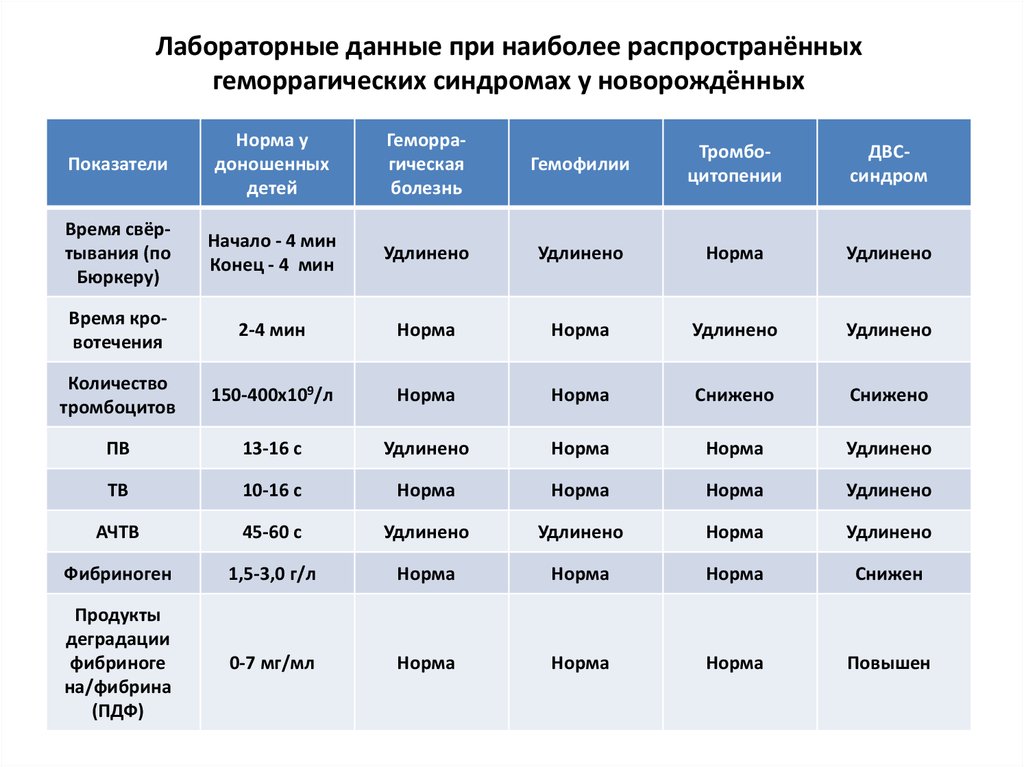 Лимфопролиферативные заболевания (лимфогрануломатоз, лимфома): характер поражения, методы верификации диагноза, основные клинические синдромы, лечение. Лимфопролиферативные заболевания. группа опухолевых заболеваний, при которых источником опухолевого роста является клетка-предшественница лимфопоэза К этой группе относятся хронический лимфолейкоз, парапротеинемические гемобластозы (множественная миелома, макро-глобулинемия Вальденстрема, болезнь тяжелых цепей). Симптомы аутоиммунного лимфопролиферативного синдрома Симптомы АЛС довольно вариабельны из-за большого количества мутаций, которые могут приводить к такому состоянию. Начало заболевания можно заметить уже на 15-й день после рождения (при врожденных формах), в детском или подростковом возрасте в случае соматических мутаций в генах FAS, CASP10 или NRAS. Обычно первым проявлением заболевания является лимфаденопатия – подмышечные, паховые или шейные лимфатические узлы увеличиваются в размерах, но при этом безболезненны и не спаяны с окружающими тканями. Регистрируется спленомегалия, в некоторых случаях она сопровождается увеличением печени (гепатоспленомегалия). Аутоиммунные проявления АЛС регистрируются обычно через некоторое время после лимфаденопатии и увеличения селезенки. В основном это поражения кровяных ростков – тромбоцитопения, гемолитическая анемия, приводящая к желтухе, изредка нейтропения. Помимо крови, аутоиммунному поражению могут подвергаться органы ЖКТ (возникают гастрит, панкреатит, колит, аутоиммунный гепатит). На коже могут проявляться признаки васкулита, делая клинику аутоиммунного лимфопролиферативного синдрома схожей с таковой при системной красной волчанке. Кроме того, могут возникать аутоиммунные формы тиреоидита, гломерулонефрита, поражаться суставы, ткани глаза (иридоциклит, увеит). Нередки поражения центральной нервной системы – эпилептические припадки, миелиты, мозжечковая атаксия. Диагностика Диагностика АЛС производится на основании осмотра, а также лабораторных, иммунологических и генетических исследований. При осмотре выявляют увеличение более чем трех групп лимфатических узлов, спленомегалию, увеличение печени. Анализ крови может показывать уменьшение количества некоторых клеток (анемию, тромбоцитопению), у части больных определяется высокая (до 30%) эозинофилия. Проба Кумбса положительная, в биохимическом анализе крови определяется выраженная гипергаммаглобулинемия. Одним из высокочувствительных методов иммунологической диагностики аутоиммунного лимфопролиферативного синдрома является проточная иммуноцитофлюориметрия, проводимая с целью выявления количества лимфоцитов с атипичным набором рецепторов (CD3+CD4-CD8-). При АЛС количество таких клеток превышает 1% от всех лимфоцитов. В биоптате лимфатических узлов определяется фолликулярная гиперплазия, результатом гистологического исследования селезенки служит лимфоидная гиперплазия. Лечение аутоиммунного лимфопролиферативного синдрома Этиотропное лечение аутоиммунного лимфопролиферативного синдрома не разработано, патогенетическая терапия сводится к применению иммуносупрессивных и цитотоксических средств. В качестве средств, подавляющих аутоиммунную активность, наиболее часто используют кортикостероиды (преднизолон, дексаметазон). К специфическим препаратам, ограничивающим скорость пролиферации лимфоцитов, относят микофенолата мофетил, сиролимус. Также при аутоиммунном лимфопролиферативном синдроме активно применяются традиционные цитотоксические средства – метотрексат, циклоспорин А и другие. При значительном увеличении селезенки или отсутствии эффекта от консервативного лечения прибегают к спленэктомии. Пересадка костного мозга и использование стволовых клеток в долгосрочной перспективе давали только временный эффект. При значительно выраженных гематологических нарушениях применяют гемотрансфузии, введение эритроцитарной или тромбоцитарной массы. Больному следует избегать физических нагрузок, использовать высоковитаминную диету. Лихорадка неясного генеза: определение, основные причины, методы диагностики, дифференциальный диагноз. Лихорадка неясного генеза (лат. febris e causa ignota) — ситуация, при которой повышение температуры тела пациента является основным или единственным симптомом, а диагноз остаётся неясным после проведения рутинного, а в ряде случаев и дополнительного обследования (то есть лихорадка неясного генеза является диагнозом исключения). В основе лихорадки неясного генеза могут лежать следующие состояния: заболевания инфекционно-воспалительного характера (генерализованные, локальные) – 30 - 50% от всех случаев (эндокардит, пиелонефрит, остеомиелит, абсцессы, туберкулез, вирусные и паразитарные инфекции и др.); онкологические заболевания - 20 – 30% (лимфома, миксома, гипернефрома, лейкемия, метастазированый рак легких, желудка и др.); системные воспаления соединительной ткани – 10 -20% (аллергический васкулит, ревматизм, ревматоидный артрит, болезнь Крона, системная красная волчанка и др.); прочие заболевания - 10 – 20% (наследственные заболевания и болезни обмена веществ, психогенные и периодические лихорадки); недиагностируемые заболевания, сопровождающиеся лихорадкой – примерно 10% (злокачественные образования, а также случаи, когда лихорадка проходит спонтанно или после применения жаропонижающих или антибактериальных средств). Механизм повышения температуры тела при заболеваниях, сопровождающихся лихорадкой, следующий: экзогенные пирогены (бактериальной и небактериальной природы) воздействуют на центр терморегуляции в гипоталамусе посредством эндогенного (лейкоцитарного, вторичного) пирогена – низкомолекулярного белка, вырабатываемого в организме. Эндогенный пироген оказывает влияние на термочувствительные нейроны гипоталамуса, приводя к резкому повышению теплопродукции в мышцах, что проявляется ознобом и снижением теплоотдачи за счет сужения сосудов кожи. Также экспериментально доказано, что различные опухоли (лимфопролиферативные опухоли, опухоли печени, почек) могут сами вырабатывать эндогенный пироген. Нарушения терморегуляции иногда могут наблюдаться при повреждениях ЦНС: кровоизлияниях, гипоталамическом синдроме, органических поражениях головного мозга. Диагностика лихорадки неясного генеза Необходимо точно соблюдать следующие критерии в постановке диагноза лихорадки неясного генеза: температура тела у пациента 38°С и выше; лихорадка (или периодические подъемы температуры) наблюдаются 3 недели и более; не определен диагноз после проведенных обследований общепринятыми методами. Пациенты с лихорадкой являются сложными для постановки диагноза. Диагностика причин лихорадки включает в себя: общий анализ крови и мочи, коагулограмму; биохимический анализ крови (сахар, АЛТ, АСТ, СРБ, сиаловые кислоты, общий белок и белковые фракции); аспириновый тест; трехчасовую термометрию; реакцию Манту; рентгенографию легких (выявление туберкулеза, саркоидоза, лимфомы, лимфогрануломатоза); ЭКГ; Эхокардиографию (исключение миксомы, эндокардита); УЗИ брюшной полости и почек; МРТ или КТ головного мозга; консультация гинеколога, невролога, ЛОР-врача. Для выявления истинных причин лихорадки одновременно с общепринятыми лабораторными анализами применяются дополнительные исследования. С этой целью назначаются: микробиологическое исследование мочи, крови, мазка из носоглотки (позволяет выявить возбудителя инфекции), анализ крови на внутриутробные инфекции; выделение вирусной культуры из секретов организма, ее ДНК, титров вирусных антител (позволяет диагностировать цитомегаловирус, токсоплазмоз, герпес, вирус Эпштейн-Барра); выявление антител к ВИЧ (метод энзим – сцепленного иммуносорбентного комплекса, тест Вестерн – блот); исследование под микроскопом толстого мазка крови (для исключения малярии); исследование крови на антинуклеарный фактор, LE-клетки (для исключения системной красной волчанки); проведение пункции костного мозга (для исключения лейкоза, лимфомы); компьютерная томография органов брюшной полости (исключение опухолевых процессов в почках и малом тазу); сцинтиграфия скелета (выявление метастазов) и денситометрия (определение плотности костной ткани) при остеомиелите, злокачественных образованиях; исследование желудочно–кишечного тракта методом лучевой диагностики, эндоскопии и биопсии (при воспалительных процессах, опухолях в кишечнике); проведение серологических реакций, в том числе реакции непрямой гемагглютинации с кишечной группой (при сальмонеллезе, бруцеллезе, болезни Лайма, тифе); сбор данных об аллергических реакциях на лекарственные препараты (при подозрении на лекарственную болезнь); изучение семейного анамнеза в плане наличия наследственных заболеваний (например, семейной средиземноморской лихорадки). Для постановки верного диагноза лихорадки могут быть повторно проведены сбор анамнеза, лабораторные исследования, которые на первом этапе могли быть ошибочными или неправильно оцененными. Апластическая анемия и агранулоцитоз: определение, основные клинико-гематологические синдромы, характер костномозгового пунктата, лечение. Апласти́ческая анеми́я — заболевание кроветворной системы, характеризуется угнетением кроветворной функции костного мозга и проявляется недостаточным образованием эритроцитов, лейкоцитов и тромбоцитов (пангемоцитопенией) или только одних эритроцитов Клиника: Анемический синдром (головокружение, снижение работоспособности, утомляемость, бледность кожных покровов и слизистых, сердцебиение, непереносимость длительных физических нагрузок и т. д.) Геморрагический синдром (кровоточивость, склонность к диапедезам, геморрагии) Инфекционные осложнения Характер пунктата: При гистологическом исследовании костного мозга обнаруживается большое количество жировой ткани, содержание которой может достигать 90 %. Среди доминирующей жировой ткани встречаются стромальные и лимфоидные элементы. Гематогенные клетки представлены крайне скудно: в небольшом количестве встречаются эритроидные и гранулоцитарные предшественники. Мегакариоциты отсутствуют Лечение апластической анемии представляет собой очень сложную задачу. Лечение с глюкокортикоидами эффективно, если болезнь обусловлена аутоиммунными механизмами, появлением антител против клеток крови. Лечение анаболическими препаратами стимулирует кроветворение. Лечение андрогенами обладает анаболическим эффектом и стимулирует эритропоэз. Лечение цитостатиками (иммунодепресантами) — назначается лишь при отсутствии эффекта от других методов лечение у больных с аутоиммунной формой, в том числе при парциальной красноклеточной аплазии. Спленэктомия Лечение антилимфоцитарным глобулином рекомендуется при отсутствии эффекта от спленэктомии и других методов лечения. Лечение циклоспорином. Циклоспорин А (сандиммун) обладает иммунодепрессантным эффектом, селективно ингибирует транскрипцию гена интерлейкина-2 в Т-лимфоцитах, подавляет продукцию Гамма интерферона и альфа фактора некроза опухоли. Трансплантация костного мозга. Агранулоцитоз представляет собой патологическое состояние, при котором наблюдается снижение уровня лейкоцитов (менее 1·109/л) за счёт гранулоцитов (менее 0,75·109/л) и моноцитов[2], повышается восприимчивость организма к бактериальным и грибковым инфекциям. Клиника: Характерны (особенно при лекарственном агранулоцитозе) острое начало и быстрое нарастание клинических симптомов. Иногда возникновению основных признаков агранулоцитоза предшествует короткий скрытый период, характеризующийся слабостью, недомоганием, головными болями[4]. К первым клиническим проявлениям агранулоцитоза относятся лихорадка, афтозный стоматит, ангина[4], озноб, артралгия[2]. Развиваются язвенно-некротические поражения слизистой оболочки полости рта в виде изъязвлений, покрытых сероватым налётом, некротических бляшек и язв с грязно-серым налётом на миндалинах (агранулоцитарная ангина)[4]. Процесс распространяется на нёбные дужки, язычок, твёрдое нёбо, дёсны и порой сопровождается незначительным увеличением регионарных лимфатических узлов, чаще всего шейных и подчелюстных. Данные изменения могут быть также на слизистой оболочке желудочно-кишечного тракта, лёгких, печени, мочевого пузыря, половых органов[4]. Лечение: В первую очередь необходимо устранение этиологического повреждающего фактора, срочная госпитализация больного агранулоцитозом, желательно в гематологическое отделение, и помещение его в стерильные условия[2] (изолятор, ультрафиолетовое облучение воздуха с защитой больного, персонал входит в масках, шапочках, бахилах). Необходим тщательный уход за кожей и слизистыми оболочками. Подкожные и внутримышечные инъекции следует заменить внутривенными, чтобы предотвратить развитие абсцессов[2]. Назначаются антибиотики широкого спектра действия, по показаниям — противогрибковые и противовирусные препараты, иммуноглобулин. Могут применяться также препараты гранулоцитарного колониестимулирующего фактора (КСФ). При некротической энтеропатии больному следует проводить парентеральное питание[2]. При аутоиммунном агранулоцитозе производится угнетение аутоиммунной агрессии, больному показаны глюкокортикоиды в высоких дозах (60—100 мг/сут) до нормализации числа гранулоцитов в крови с последующей постепенной отменой гормонов. При гаптеновом агранулоцитозе введение глюкокортикоидов не эффективно. При лечении цитостатического агранулоцитоза средствами выбора являются препараты рекомбинантных гемопоэтических Системная красная волчанка: этиология, патогенез, классификация, основные клинические проявления. Дифференциальный диагноз между системной красной волчанкой и ревматоидным артритом. Течение и исходы. Системная красная волчанка (СКВ, болезнь Либмана — Сакса; лат. lupus erythematodes, англ. systemic lupus erythematosus) — диффузное заболевание соединительной ткани, характеризующееся системным иммунокомплексным поражением соединительной ткани и её производных, с поражением сосудов микроциркуляторного русла. Системное аутоиммунное заболевание, при котором вырабатываемые иммунной системой человека антитела повреждают здоровые клетки, преимущественно повреждается соединительная ткань с обязательным наличием сосудистого компонента. Этиология: СКВ, возможно, обусловлена действием еще неизвестных триггерных факторов окружающей среды, запускающих аутоиммунные реакции у генетически предрасположенных индивидуумов КЛАССИФИКАЦИЯ Общепринятой классификации красной волчанки (КВ) не существует. Различают две основные формы болезни: дискоидную (ограниченную и Диссеминированную) и системную (острую, подострую, хроническую). При Ограниченной КВ на коже наблюдаются 1–3 очага, при диссеминированной –Свыше 3 очагов. Клиника: КЛИНИЧЕСКАЯ КАРТИНА Высыпания при ДКВ локализуются преимущественно на открытых участках тела, чаще всего – на лице, особенно на носу, щеках, лбу, ушных раковинах. Волосистая часть головы является частой, а иногда и единственной локализацией заболевания, особенно у женщин. Кожа груди и спины поражается реже. Локализация очагов на кистях является довольно редкой, особенно при их изолированном поражении Болезнь может начинаться медленно, и новые симптомы будут появляться в течение нескольких недель, месяцев или даже лет. Неспецифические жалобы на усталость и недомогание являются наиболее распространенными начальными симптомами СКВ у детей. У многих детей с СКВ отмечается периодическое или постоянное повышение температуры тела, потеря веса и аппетита. С течением времени у многих детей развиваются определенные симптомы, вызванные тем, что болезнь охватывает один или несколько органов. Очень часто заболевание затрагивает кожу и слизистые оболочки; возможны различные кожные высыпания, фотосенсибилизация (когда воздействие солнечного света вызывает сыпь) или язвы в носу либо во рту. От одной трети до половины заболевших детей имеют типичные высыпания в форме «бабочки» на носу и щеках. Иногда встречается повышенное выпадение волос (алопеция). Руки становятся красными, затем белыми, а затем синими при воздействии холода (синдром Рейно). Симптомы могут также включать опухание и тугоподвижность суставов, боли в мышцах, анемию, легкое возникновение синячков, головные боли, судороги и боль в груди. Поражение почек в той или иной степени развивается у большинства больных СКВ детей, и это главный фактор определяющий долгосрочный прогноз данного заболевания. Наиболее распространенными симптомами серьезного поражения почек являются повышение артериального давления, наличие белка, крови в моче, а также отеки, преимущественно в области стоп, голеней, а также отеки век Диагностика/диф.д: СКВ обычно можно определить по наличию повреждений на открытых участках кожи, выпадению волос, поражению слизистых оболочек рта и носа, отсутствию эрозий в суставах даже при застарелом артрите; количество лейкоцитов в синовиальной жидкости составляет < 2000 ед/мкл [2,0x109/л](преимущественно мононуклеарные клетки), выявляются антитела к двухцепочечной ДНК, заболевания почек и низкий уровень сывороточного комплемента. В отличие от РА, деформации при СКВ обычно носят обратимый характер и характеризуются отсутствием эрозий и повреждений хрящей и костей при визуализационных исследованиях. Лечение: Глюкокортикостероиды (преднизолон или др.) Терапия стволовыми клетками[10] Анти-BLyS терапия (белимумаб) Цитостатические иммунодепрессанты (азатиоприн, циклофосфан или др.) Экстракорпоральная детоксикация (плазмаферез, гемосорбция, криоплазмосорбция) Пульс-терапия высокими дозами глюкокортикостероидов и/или цитостатиков Нестероидные противовоспалительные препараты Симптоматическое лечение Моноклональные антитела: анифролумаб[en] Препараты для лечения волчаночного нефрита: белимумаб, воклоспорин[en Выживаемость через 10 лет после постановки диагноза — 90 %, через 20 лет — 87 %. Основные причины смерти: люпус-нефрит, нейро-люпус, интеркуррентные инфекции. Есть случаи выживаемости 65—87 лет. Ревматоидный артрит: этиология, патогенез, классификация, основные клинические проявления, диагностика. Дифференциальный диагноз с серонегативными спондилоартропатиями. Течение и исходы, лечение. Ревматоидный артрит (англ. Rheumatoid arthritis) — это системное воспалительное заболевание соединительной ткани с преимущественным поражением мелких суставов по типу эрозивно-деструктивного полиартрита неясной этиологии со сложным аутоиммунным патогенезом. Этиология: Как и для большинства аутоиммунных заболеваний, здесь можно выделить 3 основных фактора (ревматологическая триада): 1. Генетическая предрасположенность Наследственная склонность к аутоиммунным реакциям. Чаще встречается у носителей определённого антигена класса MHC II: HLA — DR1, DR4 2. Инфекционный фактор Гипотетические триггеры ревматических заболеваний парамиксовирусы — вирусы паротита, кори, респираторно-синцитиальной инфекции гепатовирусы — вирус гепатита B герпесвирусы — вирусы простого герпеса, опоясывающего лишая, Цитомегаловирус, вирус Эпштейна-Барр (значительно выше в синовиальной жидкости * больных РА) ретровирусы — Т-лимфотропный вирус 3. Пусковой фактор (переохлаждение, гиперинсоляция, интоксикации, мутагенные медикаменты, эндокринопатии, стрессы и т. д.). Для женщин длительность кормления грудью снижает вероятность развития РА. Кормление грудью в течение 24 месяцев и дольше понижает риск развития РА вдвое[11] Патогенез: Синовиоциты приобретают черты макрофагов, выделяют провоспалительные цитокины, в первую очередь фактор некроза опухоли альфа, интерлейкин 1, становятся антигенпрезентирующими клетками и вызывают активацию Т-хелперов 1 типа. В клетках синовиальной жидкости и в синовиальной оболочке сустава появляется большое количество Т-хелперов 1 типа, выделяющих гамма-интерферон и активирующих макрофаги. Активированные макрофаги и моноциты продуцируют провоспалительные цитокины: фактор некроза опухоли альфа, ИЛ-1, ИЛ-6. Повышение концентрации ИЛ-8 в синовиальной жидкости вызывает высокую концентрацию в ней нейтрофилов. ИЛ-1 вызывает лихорадку, активацию остеокластов, что способствует остеопорозу субхондральной пластинки кости. Фактор некроза опухоли вызывает появление молекул адгезии на поверхности эндотелиоцитов, способствуя экссудации, вызывает похудение, анемию хронического воспаления. I16, активируя гепатоциты, вызывает повышение продукции ими С-реактивного белка; активирует В-лимфоциты (превращение их в плазматические клетки). В крови значительно повышается концентрация плазматических клеток, продуцирующих иммуноглобулины. В крови и синовиальной жидкости у 80 % больных резко увеличивается концентрация IgM и IgG к изменённому Fc участку IgG (ревматоидные факторы). Выделение эндотелиального фактора роста способствует разрастанию капилляров синовиальной ткани. Ангионеогенез и пролиферация активных фибробластов, синовиоцитов приводят к образованию паннуса — агрессивной ткани, имеющей признаки опухолеподобного роста, способной внедряться в хрящ, суставную поверхность кости, образуя эрозии, и в связочный аппарат. Составляющий паннус клон неконтролируемо размножающихся, агрессивных синовиоцитов образуется сравнительно поздно — через несколько месяцев от начала заболевания. Образование иммунных комплексов в крови в результате взаимодействия IgG с ревматоидными факторами приводит к активации комплемента и повреждению микроциркуляторного русла, что объясняет висцеральные проявления ревматоидного артрита. На поздних этапах ревматоидного артрита пролиферативные процессы (рост паннуса) могут не зависеть от аутоиммунных механизмов и поддерживаются автономно. Классификация: По клиническим проявлениям (стадии) — очень ранняя: длительность до 6 месяцев; — ранняя: 6 — 12 месяцев; — развёрнутая: более года; — поздняя: более двух лет. По активности болезни (DAS28) 0 (ремиссия): DAS28 меньше 2,6; 1 (низкая): DAS28 2,6 — 3,2; 2 (средняя): DAS28 3,2 — 5,1; 3 (высокая): DAS28 больше 5,1. Инструментальная характеристика Наличие эрозии Рентгенологическая стадия (1-4) Иммунологическая характеристика Ревматоидный фактор: серо-позитивный/серо-негативный; Анти-ЦЦП: серо-позитивный/серо-негативный. По функциональным классам I сохранение самообслуживания, непрофессиональной и профессиональной деятельности II сохранение самообслуживания, не профессиональной, нарушение профессиональной деятельности III сохранение самообслуживания, нарушение профессиональной и непрофессиональной деятельности IV нарушение всех видов деятельности КЛИНИКА: Ревматоидный артрит прогрессирует в трёх стадиях. В первой стадии происходит периартикулярный отёк синовиальных сумок, вызывающий боль, местное повышение температуры и припухлость вокруг суставов. Вторая стадия — это стремительное деление клеток, которое приводит к уплотнению синовиальной оболочки. В третьей стадии воспалённые клетки высвобождают фермент, который поражает кости и хрящи, что часто приводит к деформации задетых суставов, увеличению боли и потере двигательных функций. Внесуставные проявления Со стороны сердечно-сосудистой системы: перикардит, васкулит, гранулематозное поражение клапанов, атеросклероз. Дыхательная система: плеврит, интерстициальные заболевания. Кожа: ревматоидные узелки, утолщение и гипотрофия, васкулит, сетчатое ливедо. Нервная система: компрессионная нейропатия, сенсорно-моторная нейропатия, множественные мононевриты, цервикальный миелит. Органы зрения: сухой кератоконъюктивит, эписклерит, склерит, периферическая язвенная кератопатия. Почки: амилоидоз, васкулит, нефрит, НПВП-нефропатия Кровь: анемия, тромбоцитоз, нейтропения |